21 June 2025: Articles 
Diffuse Alveolar Hemorrhage as the First Presentation of Systemic Lupus Erythematosus: A Case Report
Unusual clinical course, Challenging differential diagnosis, Management of emergency care
Selma Later ABCDEFG 1*, Muzan AbdelbagiDOI: 10.12659/AJCR.947096
Am J Case Rep 2025; 26:e947096






Abstract
BACKGROUND: Diffuse alveolar hemorrhage (DAH) is a rare but life-threatening complication of systemic lupus erythematosus (SLE), with a mortality rate of up to 80%. It is seldom the initial manifestation of SLE, making early diagnosis and treatment challenging. DAH typically presents with hemoptysis, abrupt anemia, and diffuse pulmonary infiltrates, although up to 50% of cases lack hemoptysis, complicating diagnosis.
CASE REPORT: We present a case of a 23-year-old woman with no prior SLE diagnosis who presented with dyspnea, cough, and fever. Initial investigations revealed normocytic normochromic anemia (hemoglobin 7.3 g/dL), acute kidney injury (creatinine 323 µmol/L), and bilateral pulmonary infiltrates on imaging. DAH was confirmed via bronchoscopy, which showed hemorrhagic bronchoalveolar lavage fluid, and high-resolution computed tomography of the chest revealed pulmonary consolidations with peripheral sparing. Autoimmune workup confirmed SLE, with positive antinuclear antibodies, anti-dsDNA antibodies, and low complement levels. The patient was treated with high-dose corticosteroids, rituximab, and plasma exchange. Despite initial stabilization and weaning from mechanical ventilation, she developed refractory pancytopenia, posterior reversible encephalopathy syndrome, and multi-organ failure, leading to her death on hospital day 48.
CONCLUSIONS: This case stresses the value of a high index of suspicion for DAH in young women with respiratory symptoms, even in the absence of a known SLE diagnosis. Early diagnostic measures, including bronchoscopy and imaging, are critical for timely intervention. Aggressive immunosuppressive therapy and plasma exchange can stabilize patients temporarily, but refractory cases remain challenging. A multidisciplinary approach is essential to improve outcomes in this high-mortality condition.
Keywords: Lupus Erythematosus, Systemic, Plasma Exchange, Hemorrhage, Pancytopenia, Vasculitis, Central Nervous System, Autoimmune Diseases, Immunosuppressive Agents, Lupus Vasculitis, Central Nervous System, Hemoptysis, Humans, Female, young adult, Pulmonary Alveoli, Lung Diseases, Fatal Outcome, Bronchoscopy, Tomography, X-Ray Computed
Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterized by diverse clinical manifestations and multi-organ involvement. Among its rare but life-threatening complications is diffuse alveolar hemorrhage (DAH), which occurs in less than 5% of patients with SLE and carries a high mortality rate [1,2]. DAH is particularly challenging when it presents as the initial manifestation of SLE, as its nonspecific symptoms – such as dyspnea, cough, and fever – often lead to delayed diagnosis and poor outcomes.
This case highlights the diagnostic and therapeutic challenges of DAH in a young woman with no prior SLE diagnosis. The patient’s presentation was further complicated by the absence of hemoptysis at presentation, a key symptom of DAH, and the presence of concurrent conditions, such as autoimmune hemolytic anemia and acute kidney injury. These factors emphasize the importance of maintaining a high index of suspicion for DAH in young woman presenting with respiratory symptoms, even in the absence of a known autoimmune disease.
It also highlights the importance of early diagnostic measures, such as bronchoscopy and imaging, and the role of aggressive immunosuppressive therapy in improving outcomes. Future research should focus on identifying biomarkers for early DAH detection and evaluating the efficacy of emerging therapies, such as targeted biologics, in managing this devastating complication.
Case Report
We describe a case of a 23-year-old woman who presented to the Emergency Department with dyspnea, cough, and fever for 3 days that had worsened progressively over the 3 days, leading to her seeking medical attention. On detailed history, she also reported a 6-month history of intermittent leg pain, a purpuric excoriation rash, and generalized fatigability, symptoms for which she had not sought prior medical attention.
Our patient’s past medical history was significant for anemia and a remote history of fibroids that were operated once, with recurrence. She was not known to be on long-term medications and had a negative smoking history. She denied photosensitivity, diarrhea, upper respiratory infection symptoms, night sweats, constipation, hematochezia, and hematemesis. There was no family history of autoimmune disease.
On presentation, the patient appeared pale. Her vital signs revealed tachycardia (125 beats per min), tachypnea (24 breaths per min), hypoxemia (SpO2 77% on room air), and a low-grade fever (37.5°C). Her physical examination was notable for bilateral crackles on lung auscultation. Additionally, a purpuric excoriation rash over the trunk and limbs was noted. There was no evidence of lower limb edema or signs of deep venous thrombosis.
Chest X-ray revealed bilateral perihilar diffuse opacities, which could have suggested pulmonary edema (Figure 1). Electrocardiogram revealed sinus tachycardia, with no acute ischemic changes.
Her laboratory workup on presentation is summarized in Table 1. The hematology workup was significantly deranged, with normocytic, normochromic anemia (hemoglobin of 7.3 g/dL), high reticulocytes (7.6% and an absolute count of 120×109/L), and high lactate dehydrogenase (LDH; 450 IU/L). Direct bilirubin and haptoglobin levels were normal at this point, and the direct Coombs test was positive. The blood film revealed anemia with anisocytosis. This picture was highly suggestive of autoimmune hemolytic anemia.
Kidney function tests were significant for a creatinine level of 323 μmol/L and urea level of 12 mmol/L, with no prior function tests available. The rest of the renal workup was significant for proteinuria (1.85 g/24 h), active urinary sediment was significant for 8–10 white blood cells/high-powered field and 10–12 red blood cells/high-powered field. Other striking laboratory findings included a D-dimer level of 8.1 μg/mL and severe metabolic acidosis (pH 7.1 and bicarbonate of 8.9 mmol/L). High pro-brain natriuretic peptide (BNP) was also noted, at 35 000 pg/mL). The rest of her laboratory investigations were consistent with normal levels of electrolytes and no evidence of liver injury.
Overall, the patient presented with vague respiratory symptoms, including dyspnea, cough, and fever, accompanied by type 1 respiratory failure. Given the nonspecific nature of her symptoms and the absence of hemoptysis during the initial evaluation, a broad differential diagnosis was considered. Pneumonia was high on the differentials; the presence of fever, cough, and bilateral pulmonary infiltrates on chest X-ray initially raised suspicion for pneumonia. However, the negative sputum cultures and lack of response to broad-spectrum antibiotics made this diagnosis less likely. On the other hand, the patient’s elevated pro-BNP (35 000 pg/mL) and bilateral perihilar opacities on chest X-ray could suggest cardiogenic pulmonary edema. However, echocardiography revealed only moderate pulmonary hypertension and an ejection fraction of 40%, without evidence of severe systolic or diastolic dysfunction to fully explain her respiratory failure. Furthermore, the patient’s acute kidney injury (creatinine 323 μmol/L) and proteinuria (1.85 g/24 h) raised the possibility of volume overload contributing to pulmonary edema. However, the kidney injury was more likely to be acute rather than chronic, and probably secondary rather than primary. On the other hand, elevated D-dimer levels (8.1 μg/mL) and hypoxemia prompted consideration of pulmonary embolism. Nevertheless, the absence of deep venous thrombosis on duplex ultrasound and the risk of worsening kidney function with intravenous contrast administration made the risks of CT pulmonary angiography outweigh the benefits.
Three days after admission to the hospital, the patient’s respiratory status worsened, necessitating higher oxygen requirements and non-invasive ventilation. The patient was eventually intubated and transferred to the Intensive Care Unit (ICU). Following intubation, an arterial blood gas analysis revealed a hemoglobin drop (from 7.3 to 6.0 g/dL). Despite the absence of hemoptysis, this finding, along with progressive hypoxemia and bilateral pulmonary infiltrates on chest X-ray, raised suspicion for DAH. BAL was performed, revealing blood-mixed fluid with bronchial epithelial cells, alveolar macrophages, and sparse inflammatory cells in a hemorrhagic background. Ziehl Neelsen staining was negative for acid-fast bacilli, and no malignancy was found. BAL culture grew
To investigate the underlying cause of DAH, an extensive workup was initiated. Initially, autoimmune disease was not high on the differential diagnosis, due to the patient’s nonspecific respiratory symptoms and absence of classic autoimmune features. However, after excluding other potential causes as mentioned above, a detailed autoimmune workup was pursued, which revealed findings consistent with SLE, based on the 2019 EULAR/ACR classification criteria, including positive antinuclear antibodies, anti-double-stranded DNA antibodies, low C3 and C4 complement levels, and positive lupus anticoagulant. Clinically, the patient met several criteria, such as acute kidney injury (creatinine 323 μmol/L, proteinuria 1.875 g/24 h), consistent with lupus nephritis, and autoimmune hemolytic anemia, evidenced by normocytic normochromic anemia, a positive direct Coombs test, elevated reticulocytes, high LDH, and intermittent low-grade fever (37.5–38.2°C) unexplained by infection or other causes. The absence of an alternative explanation for these findings confirmed the diagnosis of SLE as the underlying cause of DAH.
Hemoptysis was a delayed sign, occurring 10 days after admission as blood-streaked sputum. However, treatment was initiated earlier due to the high clinical suspicion of SLE, even before this symptom appeared (after clearing a urinary tract infection caused by
The patient initially responded well to treatment. Blood-stained secretions diminished gradually, and respiratory function improved over several days. Three weeks into her ICU stay, she was successfully extubated and weaned off mechanical ventilation, remaining off the ventilator for 2 weeks. She remained, however, dependent on supplemental oxygen but showed no further episodes of hemoptysis.
One week later, her overall condition deteriorated due to refractory pancytopenia, characterized by normocytic normochromic anemia, leukopenia with neutropenia, and thrombocytopenia. Further hematologic workup confirmed autoimmune hemolytic anemia, with low haptoglobin (0.2 g/L), elevated direct bilirubin (53 μmol/L), and increased transferrin saturation. These manifestations were likely a progression of her underlying autoimmune disease. Despite additional corticosteroids and PLEX therapy, her pancytopenia showed minimal improvement. Concurrently, she developed sepsis, likely exacerbated by high-dose corticosteroids and leukopenia, as well as hospital-acquired pneumonia and candidemia, which were managed according to antimicrobial sensitivity results. Although the sepsis was initially treated, it recurred, requiring intermittent hemodynamic support to maintain stability.
One month after symptoms onset, her condition was not improving, with worsening renal function, requiring renal replacement therapy, and acute respiratory distress syndrome. Additionally, she experienced an acute confusional state, accompanied by dilated pupils, sluggish light reflexes, and generalized weakness. An urgent non-contrast CT of the brain revealed bilateral occipital white matter hypodensities (Figure 3), consistent with posterior reversible encephalopathy syndrome (PRES). Given her normal blood pressure range and ongoing immunosuppressive therapy, supportive management with levetiracetam and mannitol was initiated in consultation with the neurology team.
In her final days, the patient developed melena, likely due to severe thrombocytopenia. Endoscopy revealed multiple esophageal and gastric ulcers. Despite aggressive supportive care, she progressed to multi-organ failure and severe acute respiratory distress syndrome, ultimately leading to her death on the 48th hospital day.
Discussion
SLE is a multisystem autoimmune disorder with diverse clinical manifestations, including skin rashes, arthritis, glomerulonephritis, and neurological or respiratory involvement. Among its rare but life-threatening complications is DAH, which can occasionally be the initial presentation of SLE and carries a mortality rate of up to 80% [1,2]. DAH arises from immune-mediated damage to the pulmonary microvasculature, where autoantibodies and immune complexes deposit, triggering inflammation, complement activation, and increased vascular permeability, ultimately leading to red blood cell leakage into the alveoli [2].
The differential diagnosis for DAH is broad and can be categorized into immunological and non-immunological causes. Immunological causes include autoimmune conditions such as SLE, Goodpasture syndrome, and granulomatosis with polyangiitis, while non-immunological causes encompass infections (bacterial or fungal), trauma, coagulopathies, anticoagulant overuse, pulmonary arteriovenous malformations, pulmonary emboli, and malignancies [3]. This classification aids clinicians in systematically evaluating potential etiologies and tailoring diagnostic and therapeutic approaches.
The classic triad of hemoptysis, acute anemia, and new pulmonary infiltrates is often cited as the hallmark of DAH, yet hemoptysis is absent in up to 50% of cases, making it an unreliable indicator [3]. More consistent findings include dyspnea, cough, and progressive anemia, which are present in nearly 100% of cases in some studies, alongside new pulmonary infiltrates [4]. Additional nonspecific features, such as rales, chest pain, tachypnea, and fever, often lead to initial consideration of more common conditions like pneumonia or pulmonary embolism [1,5]. In our case, the presentation was atypical, with dyspnea accompanied by elevated cardiac enzymes and D-dimer levels, segmental hypokinesia, severe renal impairment, and markers of systemic inflammation (elevated procalcitonin and C-reactive protein) and hypoperfusion (increased lactate and reduced urine output), initially suggesting alternative diagnoses such as hypertensive pulmonary edema, myocardial infarction, or pneumonia. This underscores the diagnostic challenge of DAH, particularly in the absence of typical features, and highlights the importance of maintaining a high index of suspicion in high-risk populations, such as patients with SLE. Early diagnostic measures, such as bronchoscopy with bronchoalveolar lavage (BAL) and high-resolution CT imaging, are critical for timely confirmation and intervention, as delayed diagnosis is associated with poor outcomes [6–8].
In our patient with suspected DAH, a comprehensive diagnostic pathway was used, including chest X-ray, high-resolution CT, and bronchoscopy with BAL. The latter is critical for definitive diagnosis, particularly in atypical presentations such as ours, as it typically reveals hemorrhagic fluid, with hemosiderin-laden macrophages (≥20%) serving as a key diagnostic marker. However, these macrophages may not appear until 48 to 72 h after hemorrhage, emphasizing the need for repeat testing if initial results are inconclusive [6]. Radiological imaging, including chest X-ray and high-resolution CT, was promptly conducted, revealing diffuse bilateral alveolar opacities, predominantly in the central and lower lung regions, consistent with DAH [3,4]. Despite these findings, up to 50% of DAH cases can show nonspecific or negative imaging results, underscoring the importance of integrating clinical, radiological, and bronchoscopic data for accurate diagnosis [7]. In our case, this multimodal approach was essential to confirm DAH and exclude alternative diagnoses, enabling timely initiation of life-saving therapies.
Throughout the clinical course, our patient demonstrated multiple poor prognostic markers, including the necessity for mechanical ventilation and plasmapheresis and the development of nosocomial infections, all of which are associated with increased morbidity and mortality in DAH [8]. DAH frequently correlates with heightened SLE disease activity and is commonly observed in patients with concurrent lupus nephritis, a severe manifestation of SLE. In our case, renal injury was evident, and while renal biopsy remains the criterion standard for diagnosing lupus nephritis, it was deferred due to the patient’s rapid clinical deterioration. Nonetheless, urinalysis and renal function tests strongly supported the diagnosis of lupus nephritis, further complicating the clinical picture and underscoring the aggressive nature of the disease [9]. This highlights the critical interplay between DAH and lupus nephritis, as well as the challenges in managing cases of patients with multi-organ involvement in SLE.
DAH secondary to SLE is a critical condition requiring prompt and aggressive intervention [1]. The cornerstone of initial management includes high-dose systemic glucocorticoids, administered until hemorrhage cessation, often combined with immunosuppressive agents [10]. While cyclophosphamide is frequently used, alternative therapies such as azathioprine, rituximab, and mycophenolate have been reported, although no randomized trials directly compare their efficacy [11]. In our case, mycophenolate and azathioprine were deemed more suitable for maintenance therapy than acute management. Instead, we used PLEX alongside high-dose steroids as first-line treatment, supported by evidence demonstrating the ability of PLEX to remove pathogenic immune complexes and reduce lung inflammation, particularly in refractory cases [1,10].
Adjunctive measures were also critical in our patient’s treatment. These included oxygen supplementation, coagulopathy correction, hemostasis optimization, and mechanical ventilation when necessary [12]. PLEX has emerged as a transformative therapy for severe DAH, with early intervention associated with hemorrhage control and improved pulmonary recovery [10]. Although data on PLEX remains mixed, its role as a rescue therapy in treatment-resistant cases is well-established. These advancements, coupled with a deeper understanding of SLE-related DAH, have significantly improved survival rates – from approximately 25% in the 1980s to 67% in recent years [13].
Innovative therapies like inhaled recombinant factor VIIa and inhaled tranexamic acid show promise as adjuncts in managing DAH secondary to SLE. Studies [14,15] have demonstrated their efficacy in controlling pulmonary hemorrhage in hemophilia and trauma patients, respectively, suggesting potential utility in autoimmune-related DAH, although further research is needed. These therapies offer a localized approach, minimizing systemic effects while directly addressing alveolar bleeding. However, their use must be balanced against the risk of thromboembolism, particularly in SLE patients with inherent thrombotic risks.
Incorporating inhaled recombinant factor VIIa or tranexamic acid into a multimodal strategy – alongside high-dose glucocorticoids, PLEX, and immunosuppressants – could enhance management of refractory DAH. While PLEX and steroids remain central to treatment, these innovative options provide additional tools to address severe or complex cases, reflecting advances in SLE-related DAH care. Further studies are needed to establish their safety and efficacy in autoimmune conditions; however, their potential to improve outcomes is significant.
In our case, the treatment regimen led to significant clinical and biochemical improvements, allowing the patient to be weaned off mechanical ventilation for a week. This demonstrated that PLEX provided critical stabilization, creating an opportunity for further therapies and turning a life-threatening situation into a manageable one.
Unfortunately, the patient deteriorated due to the development of additional complications. She initially presented with anemia, possibly due to chronic disease, hemorrhage, and autoimmune factors. During her ICU stay, she developed progressive autoimmune pancytopenia, further complicating diagnosis and treatment. In SLE, pancytopenia is not common and can result from hemophagocytic lymphohistiocytosis, macrophage activation syndrome, myelotoxicity, and hematological malignancies [16]. Furthermore, immunosuppressive therapies for SLE can lead to bone marrow suppression, decreasing all peripheral blood cell lineages [17]; however, the most probable culprit in our case is autoimmune hemolytic anemia (AIHA). AIHA is not uncommon in SLE, with studies estimating its prevalence at approximately 10% to 15% of SLE cases [1,2]. AIHA occurs when autoantibodies target red blood cells, leading to their premature destruction. In SLE, this is often mediated by warm-reactive IgG antibodies, although cold agglutinin disease has also been reported [3]. A large cohort study by Cervera et al found that 10% of patients with SLE developed AIHA, making it one of the more frequent hematological manifestations of the disease [4]. AIHA is often associated with high disease activity and can occur at any stage of SLE, although it is more common during flares [5]. The presence of AIHA in patients with SLE is linked to a more aggressive disease course and increased risk of other complications, such as thrombocytopenia and lupus nephritis [6].
A notable complication in our patient was PRES, diagnosed by radiological criteria. PRES, characterized by vasogenic edema in the posterior brain regions, typically presents with sudden encephalopathy or seizures and is increasingly recognized in patients with SLE [18,19]. Contributing factors in our case likely included renal impairment, fluid overload, and immunosuppressive therapy, which can exacerbate hypertension or disrupt cerebral circulation, triggering endothelial dysfunction [20,21]. While magnetic resonance imaging is the criterion standard for diagnosis, CT scans were used in our patient due to practical constraints, demonstrating their utility in such scenarios [22]. Prompt management of PRES focuses on addressing underlying causes, such as controlling blood pressure, correcting electrolyte imbalances, and treating autoimmune flares or infections. Early intervention often leads to significant symptom improvement, highlighting the importance of rapid recognition and treatment [23]. In our case, timely measures were critical in mitigating this severe complication, underscoring the need for vigilance in SLE patients with neurological symptoms.
In our case, a young SLE patient with respiratory symptoms posed a diagnostic challenge, highlighting the critical need to consider DAH, a life-threatening mimic of common conditions. Early recognition is vital, as delays can be fatal. By emphasizing rapid diagnostics like imaging or BAL, our report underscores the urgency of timely intervention, serving as a crucial alert for clinicians to act swiftly in similar high-stakes scenarios.
Conclusions
DAH is a rare but life-threatening complication of SLE. In young women with respiratory symptoms, DAH should be considered even without a prior diagnosis. Early chest imaging and diagnostic tools, such as bronchoscopy, are critical for timely detection. Prompt treatment and a multidisciplinary approach are essential to improve outcomes in these cases.
Figures
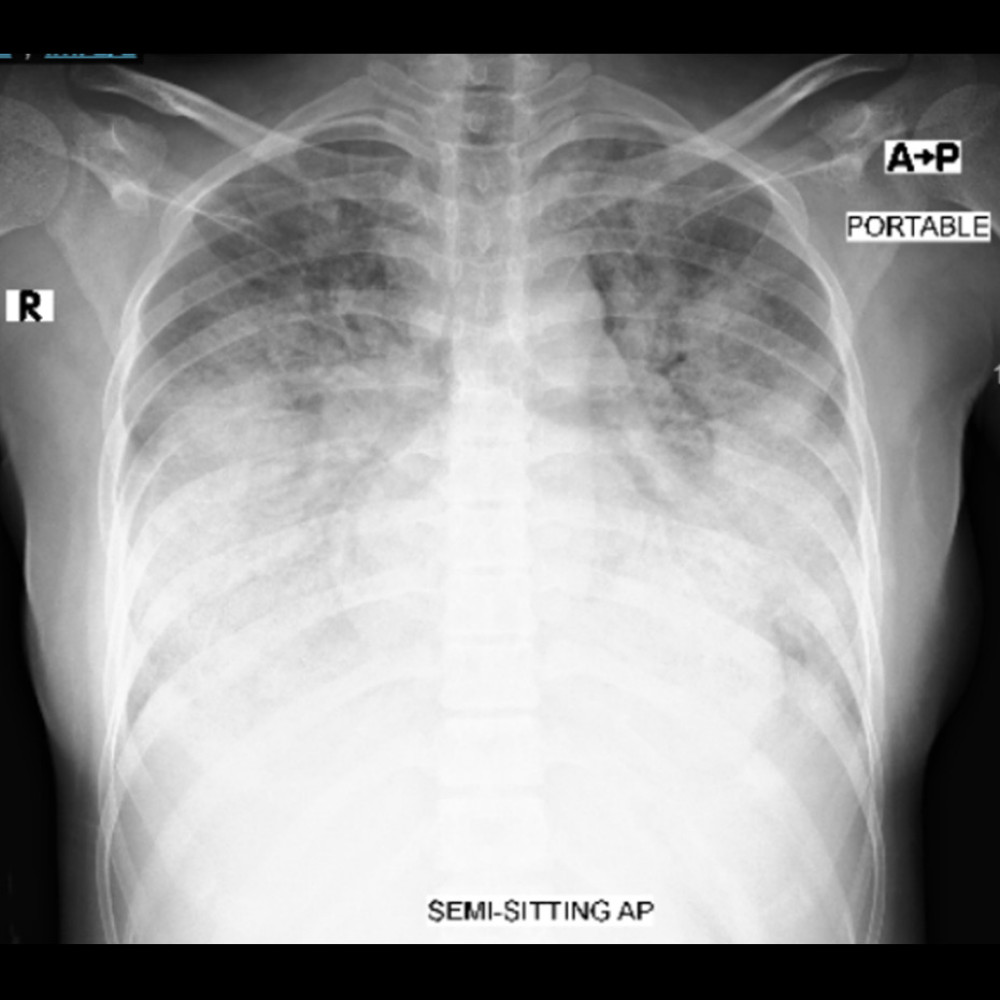 Figure 1. Chest X-ray. Anterior-posterior (AP) projection revealed bilateral diffuse airspace opacity.
Figure 1. Chest X-ray. Anterior-posterior (AP) projection revealed bilateral diffuse airspace opacity. 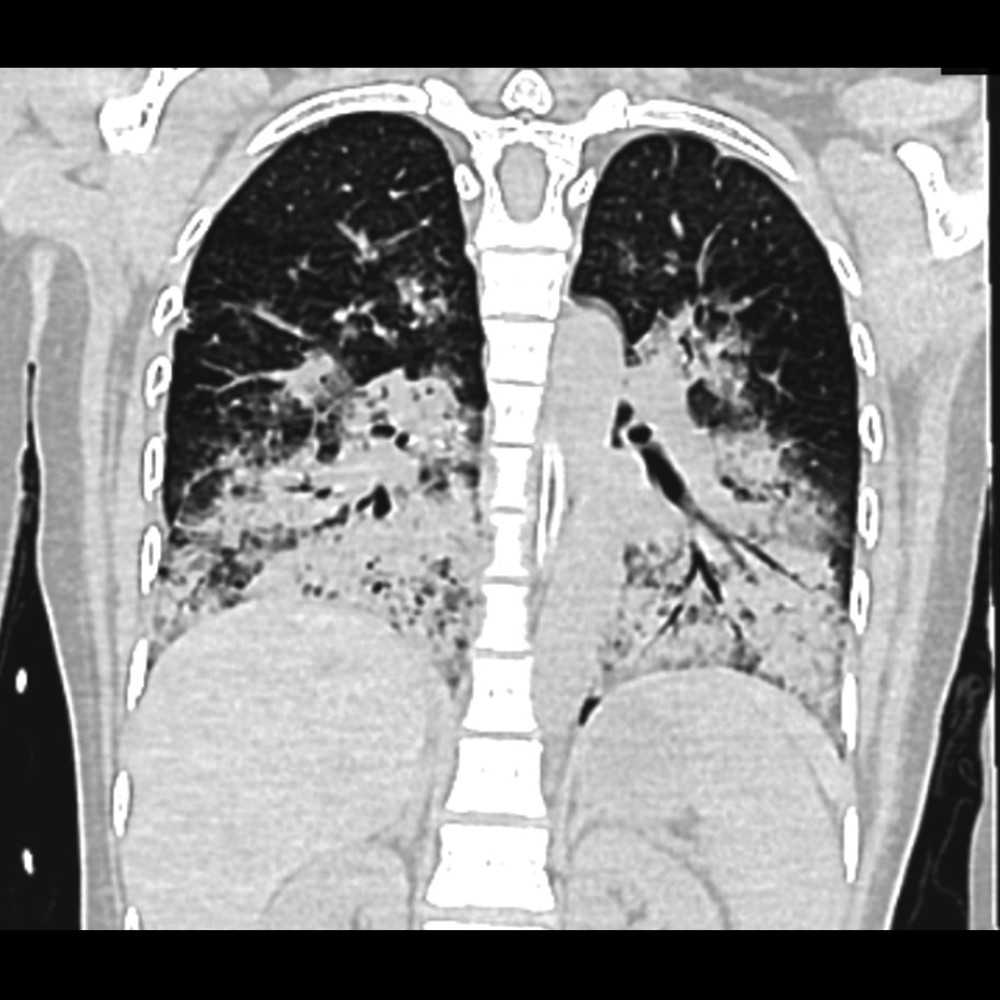 Figure 2. High-resolution computed tomography showing severe pulmonary consolidations.
Figure 2. High-resolution computed tomography showing severe pulmonary consolidations. 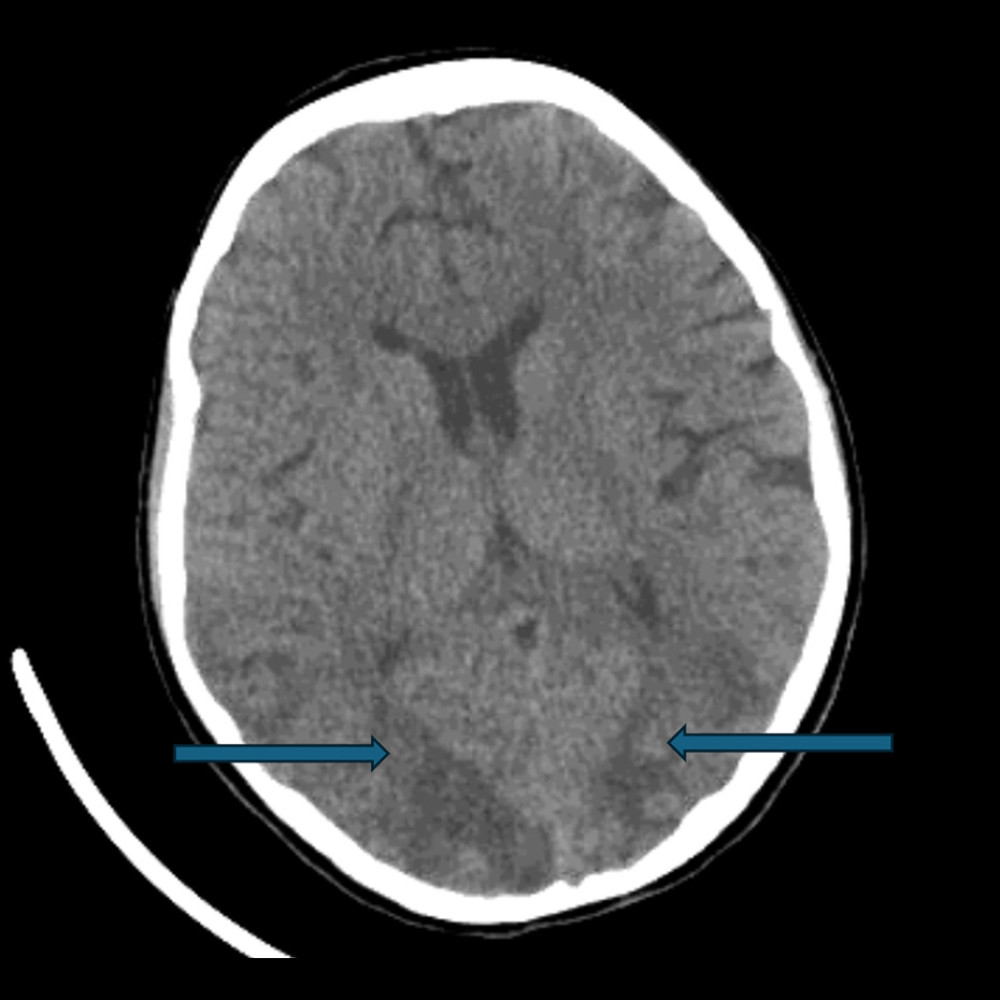 Figure 3. Computed tomography of the brain without contrast demonstrating bilateral occipital ill-defined white matter hypodensities consistent with vasogenic edema (arrows).
Figure 3. Computed tomography of the brain without contrast demonstrating bilateral occipital ill-defined white matter hypodensities consistent with vasogenic edema (arrows). 
References
1. Zamora MR, Warner ML, Tuder R, Schwarz MI, Diffuse alveolar hemorrhage and systemic lupus erythematosus: Clinical presentation, histology, survival, and outcome: Medicine, 1997; 76(3); 192-202
2. Al-Adhoubi NK, Bystrom J, Systemic lupus erythematosus and diffuse alveolar hemorrhage: Etiology and novel treatment strategies: Lupus, 2020; 29(4); 355-63
3. Park MS, Diffuse alveolar hemorrhage: Tuberc Respir Dis, 2013; 74(4); 151-62
4. Collard HR, Schwarz MI, Diffuse alveolar hemorrhage: Clin Chest Med, 2004; 25(3); 583-92
5. Aringer M, Costenbader K, Daikh D, European League Against Rheumatism (EULAR) recommendations for the management of systemic lupus erythematosus: Ann Rheum Dis, 2019; 78(6); 736-45
6. Lara AR, Schwarz MI, Diffuse alveolar hemorrhage: Chest, 2010; 137(5); 1164-71
7. Primack SL, Miller RR, Müller NL, Diffuse pulmonary hemorrhage: Clinical, pathologic, and imaging features: Am J Roentgenol, 1995; 164(2); 295-300
8. Jiang M, Chen R, Zhao L, Zhang X, Risk factors for mortality of diffuse alveolar hemorrhage in systemic lupus erythematosus: A systematic review and meta-analysis: Arthritis Res Ther, 2021; 23(1); 57
9. Ayoub I, Cassol C, Almaani S, The kidney biopsy in systemic lupus erythematosus: A view of the past and a vision of the future: Adv Chronic Kidney Dis, 2019; 26(5); 360-68
10. Fanouriakis A, Kostopoulou M, Andersen J, EULAR recommendations for the management of systemic lupus erythematosus: 2023 update: Ann Rheum Dis, 2024; 83(1); 15-29
11. Chan J, Walters GD, Puri P, Jiang SH, Safety and efficacy of biological agents in the treatment of systemic lupus erythematosus (SLE): BMC Rheumatol, 2023; 7; 37
12. Yusof MY, Bruce IN, D’Cruz D, Management and treatment of children, young people and adults with systemic lupus erythematosus: British Society for Rheumatology guideline scope: Rheumatol Adv Pract, 2023; 7(3); rkad093
13. Park JA, Treatment of diffuse alveolar hemorrhage: Controlling inflammation and obtaining rapid and effective hemostasis: Int J Mol Sci, 2021; 22(2); 793
14. Heslet L, Dalsgaard Nielsen J, Nepper-Christensen S, Local pulmonary administration of Factor VIIa (rfviia) in diffuse alveolar hemorrhage (DAH) – a review of a new treatment paradigm: Biologics, 2012; 6; 37-43
15. Solomonov A, Fruchter O, Zuckerman T, Pulmonary hemorrhage: A novel mode of therapy: Respir Med, 2009; 103(8); 1196-200
16. Levine AB, Erkan D, Clinical assessment and management of cytopenias in lupus patients: Curr Rheumatol Rep, 2011; 13(4); 291-99
17. Florez H, Morlà R, Castellanos-Moreira R, Sanmartí R, Lessons learned from bone marrow failure in systemic lupus erythematosus: Case reports and review of the literature: Semin Arthritis Rheum, 2018; 47(5); 586-92
18. Jung SM, Moon SJ, Kwok SK, Posterior reversible encephalopathy syndrome in Korean patients with systemic lupus erythematosus: Risk factors and clinical outcome: Lupus, 2013; 22(9); 885-91
19. Lai CC, Chen WS, Chang YS, Clinical features and outcomes of posterior reversible encephalopathy syndrome in patients with systemic lupus erythematosus: Arthritis Care Res, 2013; 65(11); 1766-74
20. Kagitani M, Fujiki Y, Suzuka T, A successful plasma exchange in bridging to rituximab for severe neuropsychiatric lupus and lupus nephritis with viral infections and aspiration pneumonia: Mod Rheumatol Case Rep, 2024; 8(2); 276-79
21. Dietz N, Mufti Z, Yousaf M, Acute posterior reversible encephalopathy syndrome (PRES) in the setting of interferon-beta use: BMC Neurol, 2021; 21; 445
22. Shankar J, Banfield J, Posterior reversible encephalopathy syndrome: A review: Can Assoc Radiol J, 2017; 68(2); 147-53
23. Triplett JD, Kutlubaev MA, Kermode AG, Hard T, Posterior reversible encephalopathy syndrome (PRES): Diagnosis and management: Pract Neurol, 2022; 22(3); 183-89
Figures
Figure 1. Chest X-ray. Anterior-posterior (AP) projection revealed bilateral diffuse airspace opacity.Figure 2. High-resolution computed tomography showing severe pulmonary consolidations.Figure 3. Computed tomography of the brain without contrast demonstrating bilateral occipital ill-defined white matter hypodensities consistent with vasogenic edema (arrows). In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952909
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.950868
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952931
Most Viewed Current Articles
07 Dec 2021 : Case report  22,759,422
22,759,422
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  175,936
175,936
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,499
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
65,510
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133