07 March 2021: Articles 
Kikuchi-Fujimoto Disease: Report of a Case with Progression to Lupus Nephritis
Challenging differential diagnosis, Rare coexistence of disease or pathology
Jorge Hurtado-Díaz1B, María Lucero Espinoza-Sánchez1C, Eduardo Rojas-Milán1D, Erik Cimé-Aké1E, María de los Ángeles Macias2G, Lizeth Romero-Ibarra3F, Olga Lidia Vera-Lastra14F*DOI: 10.12659/AJCR.927351
Am J Case Rep 2021; 22:e927351






Abstract
BACKGROUND: Kikuchi-Fujimoto disease (KFD) is an enigmatic disease, with a distinctive histopathology and a benign and self-limited course. It is more frequent in young Asian women. Autoimmune diseases are identified as one of its triggers; primarily SLE, which may precede, be concomitant with, or develop after the diagnosis of KFD. Patients with KFD should receive periodic follow-up for several years to detect possible evolution of SLE. The main feature of KFD is lymphadenopathy, and cervical lymph nodes are involved in 50% to 98% of cases. Other symptoms such as fever, fatigue, weight loss, and arthralgias are also reported. Differential diagnosis between KFD and SLE is a challenge. When KFD and SLE coexist, a lymph node biopsy may be diagnostic. Treatment should be symptomatic with analgesics and anti-inflammatories, with complete resolution in 3 to 4 months. Corticosteroids and immunosuppressive therapy are justified only in cases concomitant with SLE.
CASE REPORT: We report a case of KFD in a 28-year-old woman who was initially negative for anti-nuclear antibodies (ANA) and anti-double-stranded deoxyribonucleic acid antibodies (anti-dsDNA), but who became antibody-positive and presented with lupus nephritis 2 months later.
CONCLUSIONS: We present a case of a patient with KFD who developed SLE 2 months later; highlighting the importance of recognizing its association and its possible progression to monitor for future development of SLE and provide timely treatment to avoid complications. We also compared the clinical, laboratory, and histological similarities between the 2 entities.
Keywords: Histiocytic Necrotizing Lymphadenitis, Lupus Erythematosus, Systemic, Pathology, Molecular, lupus nephritis, Lymph Nodes, lymphadenopathy, Neck
Background
Kikuchi-Fujimoto disease (KFD) is a rare autoimmune-mediated entity with a benign and self-limiting course [1–4]. It presents with a higher prevalence in Asia; however, it has been reported worldwide, it is more frequent in young women and has been associated with neoplastic, genetic, infectious and autoimmune states, specifically systemic lupus erythematosus (SLE) which may precede, coexist with, or occur subsequent to the diagnosis of KFD. Patients with the initial diagnosis of KFD should be screened for SLE, and periodic follow-up is recommended to detect possible evolution of SLE [5]. Some clinical and biochemical manifestation of KFD can be shared by SLE and some diagnostic criteria of SLE can be present in KFD [6–8]. The association between KFD and SLE is rare; out of 330 cases reported between 1991 and 2005, only 28 patients with KFD met the diagnostic criteria for SLE. A Medline search revealed that, between 2005 and 2012, there were only 9 additional cases of SLE associated with KFD reported around the world [9,10]. Its association with SLE was reported by Kucukardali et al, who found SLE and KFD occurred concomitantly in 13% of cases [9]. In a systemic search that included 113 patients, SLE had been diagnosed before KFD in 18% of cases, simultaneously in 51%, and after KFD in 31%, and several cohorts with prolonged follow-up have shown that many patients develop SLE over time [11].
The main characteristics of KFD are lymphadenitis (50% to 98%), fever (30% to 67%), and headache (17% to 33%) [6]. The skin is an extra-lymphatic organ that is affected in 10% to 40% of cases [6]. The most frequent laboratory findings are increased acute-phase reactants, leukopenia with thrombocytopenia [6], and, in 7% of the cases, anti-nuclear and anti-DNA antibodies are present [2]. The histopathology finding is histiocytic necrotizing lymphadenitis. SLE is a multisystem autoimmune disease, and lymphadenopathy occurs in approximately 40% of patients, mainly in young women [7]. Differential diagnosis between KFD and SLE is a challenge because SLE closely simulates the clinical and histopathologic features of KFD. Fever and lymphadenopathy are common clinical presentations in both conditions [12], so their discrimination is based on clinical course, histopathological findings, immunohisto-chemistry, and determination of antibodies [12]. Treatment is symptomatic, with analgesics and anti-inflammatories [6,7]. Corticoids and immunosuppressive therapy are justified in cases with concomitant SLE.
The aim of this report is to present the case of a 28-year-old woman with KFD who developed lupus nephritis 2 months later. We highlight its association and the importance of follow-up to detect possible progression, and compare the clinical, laboratory, and histologic similarities between these 2 entities.
Case Report
The patient was a 28-year-old woman who did not have a family or personal history of autoimmune disease. Her illness started 1 month before her hospital admission, with fever of 39°C, predominantly nocturnal, accompanied by sweating, fatigue, severe headache, anxiety, and agitation. She had weight loss of 10 kg, accompanied by decreased appetite, nausea, and foul-smelling, greenish, loose stools without mucus or blood.
A physical examination on admission showed hypotension, tachycardia, polypnea, and fever of 39°C. She was conscious, agitated, talkative, and anxious. She had alopecia and her face had papules, pustules, vesicles with hypopigmentation, meliceric crusts, and lichenification (Figure 1A). The oral cavity had red, non-painful ulcers, candidiasis, and painful bilateral parotid hypertrophy. The neck had left-side adenopathy fixed to deep planes of 2×2 cm, of hard consistency, painful on palpation (Figure 1B), without airway compromise. Precordial areas were without alterations, and she had bilateral pleural effusion syndrome of approximately 40%. The right axillary had adenomegaly of 1 cm of rubbery consistency not fixed to deep planes and painful. Her abdomen was soft and depressible, without signs of rebound tenderness. There was hepatomegaly without splenomegaly. The genital area had transvaginal bleeding. She had edema in lower extremities, normal pulses, and reflexes, and no arthritis.
Initial laboratory tests (Table 1) showed acute kidney involvement, cholestatic hyperbilirubinemia, hepatocellular affection, and pancytopenia, with negative antinuclear and anti-DNA antibodies, hypocomplementemia, elevation of acute-phase reactants, negative viral panel and TORCH, urine culture and blood culture without bacterial growth. Plain neck tomography was performed, showing right cervical adenomegalies, forming conglomerates radiologically diffuse and heterogeneous (Figure 1B). Ultrasound of the liver and bile ducts showed grade II hepatic steatosis and spleen with normal size and echogenicity. Suspecting a lymphoproliferative syndrome, bone marrow aspirate was performed with normal cellularity for age. Excisional lymph node biopsy reported necrotizing histiocytic lymphadenitis and immunohistochemistry with the streptavidinbiotin system showed presence of CD 68-positive and CD 123-negative in macrophage cells (Figure 1C, 1D); therefore, KFD was diagnosed.
After ruling out infectious, lymphoproliferative, and autoimmune processes such as SLE, she received symptomatic treatment with paracetamol and naproxen. She presented complete clinical and biochemical remission during a period of 60 days. Later, a stormy course was observed, with appearance of renal involvement (Table 1). Given the high suspicion of lupus nephritis, the renal biopsy reported class III lupus nephropathy and an immunological profile consistent with positive ANA (1: 160) and anti-dsDNA, hypocomplementemia (C3 29 mg/dl, C4 6 mg/dl), and lymphopenia (500 109/L). The patient met the American College of Rheumatology criteria for SLE. Therefore, induction to remission was initiated with pulses of methylprednisolone at a dose of 1 g/day for 3 days and cyclophosphamide with a cumulative dose of 7.1 g. The maintenance therapy was performed with prednisone, mycophenolate mofetil, and chloroquine. By the eighth month of treatment, the glomerular filtration rate and serum creatinine normalized with a decrease of proteinuria and recovery of serum albumin and the other laboratory parameters (Table 1). After 2 years of follow-up, she remains in remission.
Discussion
The etiology of KFD remains unknown, although a viral or autoimmune pathogenesis, such as systemic lupus erythematosus (SLE), has been suggested [13]. The exact physio-pathological relationship between these 2 diseases is still unclear [14]. Kucukardali et al reported 28 cases of SLE-associated KDF that met the diagnostic criteria for SLE; of those, only 6 were diagnosed with SLE after KFD [9]. Santana et al reported a total of 35 cases of SLE-associated KFD, in which 14 patients were diagnosed with SLE after a diagnosis of KFD [15]. Goldblatt et al reported 4 cases in which KFD preceded a clinical and immunological diagnosis of SLE by between 3 and 14 months [5] and Patra et al reported a case of a woman who developed SLE 2 years after KFD [16]. Sopeña et al reported that only 25 of 113 patients with KFD-SLE had lupus nephritis, and of those, only 8 develop lupus nephritis after KFD. Therefore, in patients with KFD-SLE, the prevalence of lupus nephritis is lower in patients with classic SLE [11]. In our case, KFD was present before lupus nephritis, and the time of progression was 2 months. Some authors have postulated that KFD is prodromal for SLE, and this is supported by the fact that microscopic features of KFD can be similar to those found in lupus lymphadenitis [4]; therefore, periodic follow-up is recommended to detect possible evolution of SLE [5].
Our patient had common symptoms and laboratory findings of KFD, such as fever, fatigue, arthralgias, pancytopenia, especially severe anemia, increased erythrocyte sedimentation rate (ESR) at the beginning, and these can be found in both entities (Table 2). Our patient also had diarrhea, which is rarely reported in KFD. In this regard,
KFD skin lesions are nonspecific (eg, macules, papules, plaques, facial malar erythema, erosions, patches, and nodules) and have been observed in up to 40% of cases and may be similar to those observed in lupus. Kim et al found that SLE-KFD patients tend to have high incidence of skin manifestations [19], and patients with KFD and skin lesions can develop SLE [20]. Therefore, clinical features of SLE and KFD can be similar; nevertheless, definite discrimination between those 2 diseases is based on histopathological findings. Histopathologic features that support SLE include an increased number of plasma cells, hematoxylin bodies, DNA deposits in the vascular walls, neutrophilic infiltration, and varying degrees of coagulative necrosis with Azzopardi phenomenon [20]. The histopathology of KFD is sufficiently distinctive to allow its recognition as a specific entity [11,17,20]; the absence of hematoxylin bodies and neutrophils and CD68+ histiocyte infiltrate indicate KFD rather than SLE, and our patient showed definite features of KFD. With respect to autoantibodies, it is important to note that in KFD they are only present in 7% of cases [17,20] whereas in SLE they are positive in over than 98% of cases. In KFD, laboratory findings are nonspecific, including elevated ESR, neutropenia, lymphocytosis, mildly elevated transaminase, elevated LDH and ANA, and reduced C3 values. These findings were observed in our patient, and have been reported in the literature [10]. Table 2 provides a comparison of clinical, laboratory, and histological findings between KFD and SLE.
Regarding treatment of KFD, 64 to 80% of cases do not require specific therapy, whereas SLE patients need immunosuppressive management to modify its course and prognosis. In KFD, treatment is symptomatic with nonsteroidal anti-inflammatory drugs (NSAIDs) with a short course of steroids. When KFD coexists with SLE, it can show a more aggressive course, and the treatment is aimed to prevent relapse [11]. After 2 years of follow-up, our patient is in remission of lupus nephritis and she remains in disease remission.
The presence of histiocytic necrotizing lymphadenitis, the absence of ANAs, double-stranded DNA antibodies, and the favorable clinical response with symptomatic management allowed us to rule out the diagnosis of SLE at the beginning of the disease. Subsequently, recurrence, stormy course, presence of pancytopenia, hypocomplementemia, erythematous skin lesions, and rapid and progressive deterioration of renal function confirmed by renal biopsy substantiated the diagnosis of SLE.
Conclusions
We present the case of a 28-year-old woman with KFD who was initially negative for antibodies and developed SLE 2 months later. Differential diagnosis between these 2 entities is difficult due to the similar spectrum of clinical manifestations; therefore, biopsy, immunohistochemistry, and antibody determination are necessary. We emphasize the importance of recognizing the association of these 2 entities and possible progression to provide timely treatment and avoid complications.
References:
1.. Bosch X, Guilabert A, Miquel R, Campo E, Enigmatic Kikuchi-Fujimoto disease: A comprehensive review: Am J Clin Pathol, 2004; 122; 141-52
2.. Bosch X, Guilabert A, Kikuchi-Fujimoto disease: Orphanet J Rare Dis, 2006; 1; 18
3.. Hu S, Kuo T-T, Hong H-S, Lupus lymphadenitis simulating Kikuchi’s lymphadenitis in patients with systemic lupus erythematosus: A clinicopathological analysis of six cases and review of the literature: Pathol Int, 2003; 53; 221-26
4.. Quintas-Cardama A, Fraga M, Cozzi SN, Fatal Kikuchi-Fujimoto disease. The lupus connection: Ann Hematol, 2003; 82(3); 186-88
5.. Goldblatt F, Andrews J, Russell A, Isenberg D, Association of Kikuchi-Fujimoto’s disease with SLE: Rheumatology, 2008; 47(4); 553-54
6.. Dumas G, Prendki V, Haroche J, Kikuchi-Fujimoto disease: Retrospective study of 91 cases and review of the literature: Medicine (Baltimore), 2014; 93(24); 372-82
7.. Astudillo L, [Kikuchi-Fujimoto disease]: Rev Med Interne, 2010; 31(11); 757-65 [in French]
8.. Khanna D, Shrivastava A, Malur PR, Kangle R, Necrotizing lymphadenitis in systemic lupus erythematosus: Is it kikuchi-fujimoto disease?: J Clin Rheumatol, 2010; 16(3); 123-24
9.. Kucukardali Y, Solmazgul E, Kunter E, Kikuchi-Fujimoto disease: Analysis of 244 cases: Clin Rheumatol, 2007; 26(1); 50-54
10.. Zuo Y, Foshat M, Qian YW, A rare case of Kikuchi Fujimoto’s disease with subsequent development of systemic lupus erythematosus: Case Rep Rheumato, 2012; 2012; 325062
11.. Sopeña B, Rivera A, Chamorro A, Clinical association between Kikuchi’s disease and systemic lupus erythematosus: A systematic literature review: Semin Arthritis Rheum, 2017; 47(1); 46-52
12.. Salamat S, Chan J, Jolly K, Kikuchi-Fujimoto disease and prognostic implications: Head Neck Pathol, 2020; 14(1); 272-75
13.. Petri M, Orbai AM, Alarcón GS, Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus: Arthritis Rheum, 2012; 64(8); 2677-86
14.. Londhey VA, Buche AS, Kini SH, Rajadhyaksha GC, Kikuchi Fujimoto disease and systemic lupus erythematosus – a rare association: J Assoc Physicians India, 2010; 58; 642-43
15.. Santana A, Lessa B, Galrão L, Kikuchi-Fujimoto’s disease associated with systemic lupus erythematosus: Case report and review of the literature: Clin Rheumatol, 2005; 24(1); 60-63
16.. Patra A, Bhattacharya SK, SLE developing in a follow-up patient of Kikuchi’s disease: A rare disorder: J Clin Diagnostic Res, 2013; 7(4); 752-53
17.. Liatsos GD, Pirounaki M, Skounakis M, Moulakakis A, Novel presentation of Kikuchi-Fujimoto disease with chronic, febrile diarrhoea, mimicking inflammatory bowel disease: Ann Hematol, 2007; 86(10); 773-75
18.. Charalabopoulos K, Charalabopoulos A, Papadopoulou CH, Papalimneou V, Giardia lamblia intestinalis: A new pathogen with possible link to Kikuchi-Fujimoto disease. An additional element in the disease jigsaw: Int J Clin Pract, 2004; 58(12); 1180-83
19.. Aydogan T, Kanbay M, Uraldi C, Kikuchi Fujimoto disease secondary to Entamoeba histolytica: Case report: J Infect, 2006; 53(4); e171-73
20.. Găman M, Vlădăreanu AM, Dobrea C, A challenging case of Kikuchi-Fujimoto disease associated with systemic lupus erythematosus and review of the literature: Case Rep Hematol, 2018; 2018; 1791627
Tables
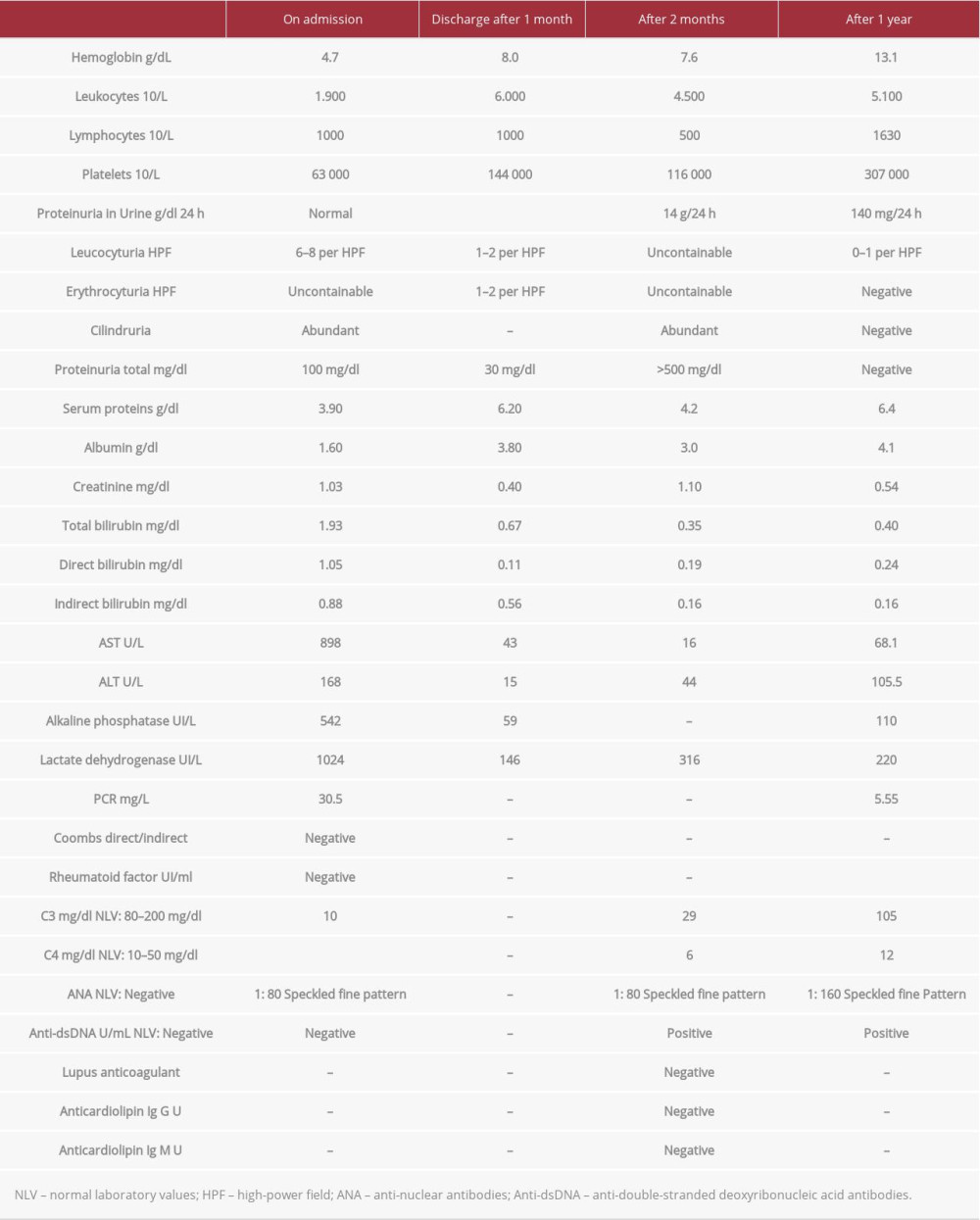 Table 1.. Evolution of laboratory finding. On admission, discharge after 1 month, 2 months and 1 year of evolution.
Table 1.. Evolution of laboratory finding. On admission, discharge after 1 month, 2 months and 1 year of evolution.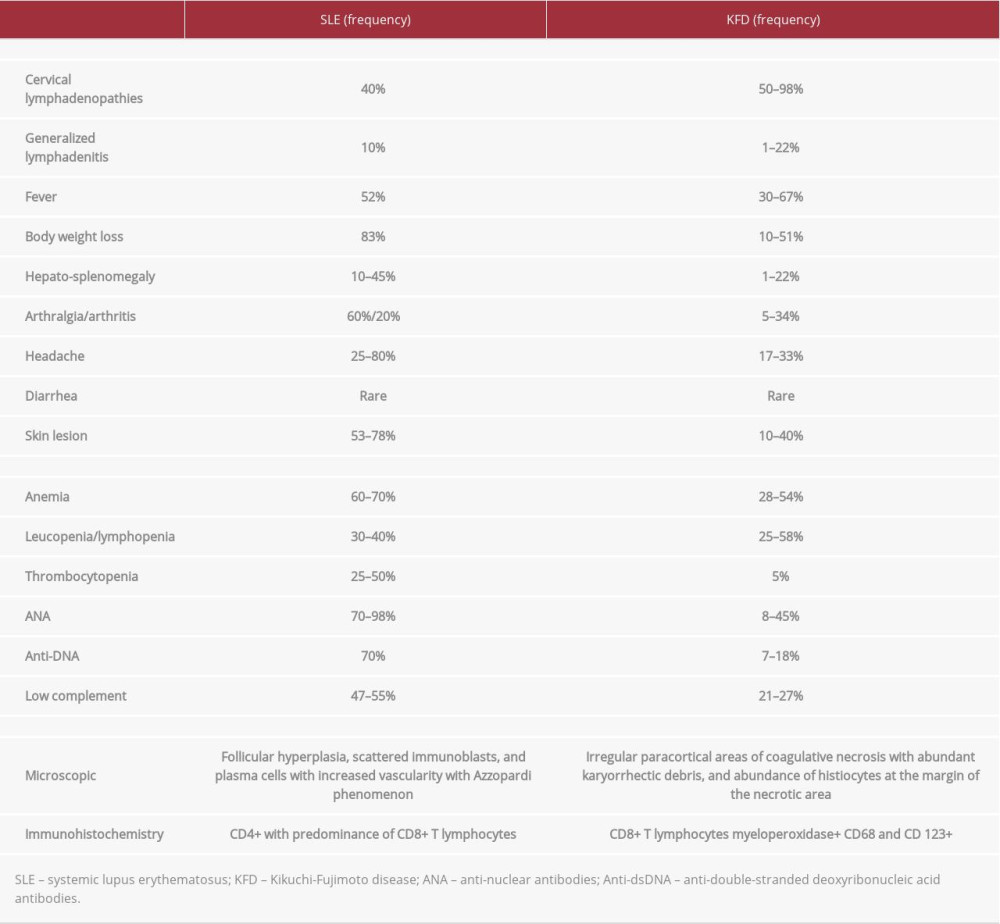 Table 2.. Comparison among clinical, laboratory, and histological findings in SLE and KFD.Table 1.. Evolution of laboratory finding. On admission, discharge after 1 month, 2 months and 1 year of evolution.Table 2.. Comparison among clinical, laboratory, and histological findings in SLE and KFD.
Table 2.. Comparison among clinical, laboratory, and histological findings in SLE and KFD.Table 1.. Evolution of laboratory finding. On admission, discharge after 1 month, 2 months and 1 year of evolution.Table 2.. Comparison among clinical, laboratory, and histological findings in SLE and KFD. In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953068
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133