08 December 2021: Articles 
A Case Series of Adult Patients Diagnosed with IgA Vasculitis Requiring Systemic Immunosuppression
Unusual setting of medical care, Rare disease, Adverse events of drug therapy
Fenfen Cai1ABCDEF*, Lisa Phipps2AE, Peter Wu3AE, Ming-Wei Lin145ABCDEFGDOI: 10.12659/AJCR.933407
Am J Case Rep 2021; 22:e933407






Abstract
BACKGROUND: IgA vasculitis (IgAV) is a rare and potentially life-threatening small-vessel vasculitis in adults. The disease course is often more severe than its childhood counterpart. The disease is noted for its heterogeneous presentation with varying severity. There are no current treatment guidelines for severe multi-organ involvement of IgAV. The treatment approaches based on the clinical discretion of treating doctors remain controversial, especially regarding the role, duration, and type of immunosuppression.
CASE REPORT: We present 3 cases of severe multi-organ IgAV encountered at our tertiary referral center between 2016 and 2021, which were treated with different immunosuppression regimens, including combination of systemic corticosteroids, oral immunosuppressants (azathioprine, mycophenolate, and sirolimus), rituximab, and cyclophosphamide. In these patients, IgAV presented differently but were all organ-threatening or life-threatening in nature. IgAV in all patients responded to therapies; however, infection complicating underlying comorbidities was the cause of death in 1 patient and the cause of comorbidities in the other 2. Other treatment-related complications included weight gain, adrenal insufficiency, and secondary hypogammaglobulinemia.
CONCLUSIONS: IgAV can be a polyphasic and a potentially challenging severe organ-threatening disease to treat in adults. The outcomes presented here highlight the morbidity and substantial risks involved in treating complex IgAV patients. Early use of biologics may have a role in preventing treatment-related toxicity. Further studies on IgAV in adults are urgently needed.
Keywords: Cyclophosphamide, Immunoglobulin A, rituximab, Vasculitis, Child, Humans, IgA Vasculitis, Immunosuppression Therapy
Background
IgA vasculitis (IgAV) is an immune complex medicated small-vessel leukocytoclastic vasculitis that is common in children but rare in adults [1]. While it is often benign in childhood, in adults it has a more severe course and is known to be associated with increased risk of significant renal impairment. Due to its rarity, the understanding of its pathogenesis, natural history, and disease course in adults is limited. There is also no consensus in the management of IgAV in patients with severe multi-organ involvement. The management often involves complex and long-term systemic immunosuppression. Randomized controlled studies that could inform therapy guidelines are difficult to conduct due to the low incidence.
We describe 3 cases of severe manifestations of IgA vasculitis in adult patients at our tertiary referral center between 2016 and 2021 treated with intensive systemic immunosuppression (Table 1). Systemic immunosuppression including corticosteroids, azathioprine, mycophenolate, sirolimus, rituximab, cyclophosphamide was used to achieve control of the disease. We discuss a personalized, stratified treatment approach, treatment response, and outcome in these patients after more than 12 months of follow-up.
Case Reports
CASE 1:
A 31-year-old Aboriginal woman with a strong family history of systemic lupus erythematosus presented with purpuric bullous skin eruptions on the lower limbs 20 days after recovering from a bout of tonsillitis (ASOT-positive) (Figure 1). She then developed concomitant polyarthritis of hands, knees, shoulders, and ankles, and significant abdominal pain and diarrhea. Iron-deficiency anemia with a hemoglobin of 72 g/L, proteinuria (protein output 2.72 g/L/24 h, albumin/creatinine ratio 112.7 mg/mmol) and hematuria (>100×10E6/L) were also noted. Her creatinine remained normal. She was commenced on prednisolone at a dose between 40–100 mg daily (body weight 105 kg) for 2 weeks before her transfer to our tertiary referral center.
Her autoimmune serology for antinuclear antibodies (ANA), extractable nuclear antibodies (ENA) and antineutrophil cytoplasmic antibodies (ANCA), rheumatoid factor (RhF), anticyclic citrullinated peptide antibodies (CCP), and antiphospholipid antibodies (including anticardiolipin IgG, lupus anticoagulant, and anti-β2glycoprotein 1) was not detected. Colonoscopy and endoscopy revealed shallow ulcers and active ileitis and colitis at the ileocecal valve. Her skin biopsies showed features of leukocytoclastic vasculitis. A kidney biopsy confirmed the presence of crescentic glomerulonephritis with immunodeposits, including trace, patchy, granular, and mesangiocapillary IgA, which may have been attenuated by the preceding weeks of corticosteroid therapy (Figure 2). A diagnosis of crescentic IgAV nephritis was thought to be most likely, corresponding to her IgA leukocytoclastic vasculitis.
The main affected organs were the kidneys and skin. Immunosuppression was deemed particularly pertinent to maintain renal function. Oral prednisolone was continued at 80 mg daily while other steroid-sparing systemic immuno-suppression was introduced. Cyclophosphamide (EuroLupus protocol of 500 mg every 2 weeks) was commenced and continued for a total of 12 cycles for persisting proteinuria and recurrence of new skin lesions. The decision to use cyclophosphamide was based on our experience of cyclophosphamide in the treatment of vasculitic glomerulonephritis. On histology, there was evidence of crescents in 8 out of 20 glomeruli seen, 4 of which displayed segmental sclerosis. The light microscopy findings were consistent with ISKDC Class IIIa (focal mesangial proliferation with <50% crescents). Following induction of remission, mycophenolate was added for remission maintenance. Her hematological and biochemical parameters, inflammatory markers, urinary indices, and treatment-related complications were carefully monitored throughout her treatment, which was adjusted according to her clinical progress. Her fasting blood sugar level and bone mineral density were normal. Management for her significant weight gain of 20 kg, bone health, and post-cyclophosphamide surveillance are ongoing. Twelve months after initiation of treatment, the patient remains on mycophenolate 1.5 g twice daily, and low-dose prednisolone (0.1 mg/kg/day). There was normalization of inflammatory markers and a significant reduction in her albumin/ creatinine ratio (5.6 mg/mmol), improvement in her hemoglobin (116 g/L), and reduction in the frequency of skin lesion recurrence. The patient had several episodes of urinary tract infections, which were successfully treated with oral antibiotics.
CASE 2:
A 43-year-old Indian man presented with a 2-week history of progressive constant generalized abdominal pain and a 1-week history of petechiae, macular palpable lesions over dorsum feet, legs, hands, and abdomen. His abdominal pain was not complicated by melena or hematemesis. He did not have documented hematuria or proteinuria.
A skin biopsy revealed evidence of granular deposition of IgA and C3 in the superficial dermal vessels, a pattern consistent with IgA vasculitis. CT abdomen demonstrated small-bowel enteritis. Enteroscopic findings were consistent with macroscopic vasculitis involving the jejunum and duodenum (Figure 3). A diagnosis of IgA vasculitis was made after the exclusion of other causes of systemic vasculitis. Due to his significant abdominal symptoms, the gastrointestinal (GI) IgAV required systemic immunosuppression. He was commenced on systemic immunosuppression with oral prednisolone 75 mg (body weight 107 kg) and azathioprine based on management of other similar autoimmune colitis, namely Crohn’s disease and ulcerative colitis. His vasculitis was well suppressed and entered serological remission with a weaning dose of predniso-lone and maximally tolerated dose of azathioprine at 150 mg daily. His weight gain, metabolic profile, and bone health remain under ongoing monitoring. The patient had viral respiratory tract infections, which were self-limiting.
CASE 3:
A 64-year-old Turkish woman initially presented with petechial, ecchymoses, and palpable purpuric skin lesions over the lower limbs and abdomen. Her skin biopsy was consistent with IgA vasculitis. Mesenteric involvement was suspected due to intermittent abdominal pain and reduction in hemoglobulin. Hemoptysis and ground-glass appearance on CT of the chest (Figure 4) were investigated with a lung biopsy, which demonstrated capillaritis suggestive of vasculitis pulmonary involvement. There was no evidence of renal involvement. The patient had cardiomyopathy, pulmonary hypertension, valvular disease, and pre-existing stage 3 chronic kidney disease as co-mobilities.
Systemic immunosuppression was necessary to induce clinical remission of her pulmonary, cutaneous, and GI vasculitis. Of these, pulmonary manifestations were particularly concerning as she had presented on multiple occasions with life-threatening hemoptysis. She was treated with a weaning dose of oral prednisolone and azathioprine at 150 mg daily. The patient did not have further hemoptysis but continued to have recurrent skin lesions. Another steroid-sparing agent, mycophenolate, was trialed, but her progress was hindered by recurrent sepsis. Due to a history of cardiomyopathy and neutropenia secondary to valganciclovir, which was used to treat low-level CMV reactivation, cyclophosphamide was contraindicated. The patient had multiple admissions due to disease relapses, mostly in the form of recurring skin lesions and a drop in hemoglobin. These relapses were treated with courses of pulsed methylprednisolone (1 g daily for 3 days) with follow-on oral prednisolone. Based on case reports on the use of B-cell-depleting therapy in refractory IgAV [2], Rituximab (intravenous, 1 g on day 0 and day 14) for induction of remission was also given. Sirolimus as a steroid-sparing agent was used in combination with a low dose of prednisolone. The patient developed adrenal insufficiency and secondary hypogammaglobulinemia, which was treated with monthly intravenous immunoglobulin replacement therapy. Her vasculitis entered clinical remission after rituximab, and with sirolimus and prednisolone combination, and prednisolone was ultimately able to be weaned off completely without any relapse of her disease.
The patient, unfortunately, did not survive her other comorbidities, including cardiomyopathy, severe pulmonary hyper-tension, valvular disease, multifactorial chronic kidney disease, and aspiration pneumonia. She died 4 years after the diagnosis of IgA vasculitis. At the time of her death and in the preceding year leading up to her death, her IgA vasculitis was in complete remission while she was off steroids.
Discussion
In adults, the annual incidence of IgA vasculitis (IgAV) is 0.8–2.2 per 100 000 individuals [3]. The disease is more common in males (male/female ratio of 1.5) [3]. The associations of IgAV with hypertension, diabetes, and obesity were observed [4]. The deposition of IgA1-dominant immune complexes is thought to be central to the pathogenesis of the disease [5]. Specific antigen exposure in the mucosa of genetically predisposed individuals could be associated with increased IgA1 synthesis. An aberrant glycosylation of IgA1 leads to the production and deposition of immune complexes [5]. Rarely, malignancies (especially solid malignancies) had been linked to IgAV [6]. However, epidemiological studies addressing the malignancy association are scarce. It is known that HLA-DRB1, mainly HLA-DR1*0103, is strongly associated with IgAV susceptibility but its association with ethnicity is unknown [7]. Further large studies in adults with IgAV are needed in the search for genetic markers of disease susceptibility, disease severity, and prognosis.
The clinical manifestations of IgAV in adults can vary significantly from its monophasic childhood presentation. The common spectrum of disease includes cutaneous purpura, arthralgia (61%), acute enteritis (48–53%) and glomerulonephritis (45–85%) [4,8,9]. Symmetric palpable purpura is nearly constant in patients [8]. In 1 cohort, necrotic and bullous lesions developed in 45.5% of all patients [1]. In our patients, severe cutaneous involvement, including necrotic/bullous lesions, joint disease, enteritis, renal involvement, and a rare complication of pulmonary involvement, was present. Predictors for disease severity are not well studied [1]. It is known that pre-existing comorbidities predict premature death [10].
There are significant uncertainties with respect to the treatment and prognosis of severe organ-threatening IgAV in adults. While mild to moderate cutaneous and/or articular forms of IgAV are mostly managed symptomatically, severe visceral organ manifestations require complex and long-term immuno-suppression [8]. Renal complications are a major determinant of long-term morbidity and mortality of IgAV [7]. In patients with kidney involvement, 11% will progress to end-stage renal disease (ESRD) [9]. Poor prognosis factors are impaired renal function at baseline, proteinuria >1 g/day at disease onset, macroscopic hematuria, hypertension, and persistent proteinuria >1 g/day during follow-up [9]. Up to 50% of such patients will progress of ESRD over 10 years. Cyclophosphamide has been the standard induction treatment for renal vasculitis, including IgA nephritis [8,11]. There are no specific treatment guidelines in adults. Kidney Disease Improving Global Outcomes (KDIGO) and European treatment guidelines provide references for IgAV nephritis in children [12,13]. Both recommend a treatment strategy of cyclophosphamide and steroids for severe disease [14]. Despite some success in case reports, the role of rituximab for induction of remission is controversial in IgAV renal disease. A randomized controlled trial of 34 patients at high risk of renal progression compared a standard-care group treated with diet, renin-angiotensin-aldosterone system blockade, and fish oil, and a group that received rituximab. Rituximab failed to significantly reduce proteinuria or benefit renal function despite effective CD 19+ B cell depletion [15]. Ren et al examined mycophenolate mofetil with low-dose prednisolone in patients with IgA nephritis with large proteinuria (>2.0 g/24 h). At 6 months, there was no significant difference in the remission rate between the mycopheno-late and the full-dose prednisolone (0.8–1.0 mg/kg/day) group. This supports the role of mycophenolate as a steroid-sparing agent [8]. Predicting long-term renal outcome is however limited by the lack of prospective, randomized controlled trials to analyze the benefit-risk ratio of long-term immunosuppression in this setting [8].
Abdominal pain, purpura, and arthralgia form the “classic triad” of IgAV. It was present in all our patients. Gastrointestinal symptoms are caused by bowel ischemia and edema. Serious complications include intussusception, infarction, and perforation, requiring surgical management. There is no standardized treatment protocol for IgAV GI manifestations in adults for induction of remission or as maintenance therapy. Oshikata et al reported a case of catastrophic IgAV with non-occlusive mesenteric ischemia causing death despite aggressive immuno-suppression, including IV steroids and cyclophosphamide [16]. Early use of prednisone at 1 to 2 mg/kg/day for 1 to 2 weeks before tapering was effective in reducing abdominal pain and can prevent GI bleeding or intussusception in children [17,18]. Our second case, due to the refractory nature of skin lesions and abdominal symptoms, required long-term immunosuppression. The successful treatment with azathioprine led to disease remission and a complete cessation of prednisolone by 6 months.
Early treatment of our third patient was marred by treatment-related complications and/or inadequate response. Rituximab was reviewed to be a safe and effective agent in treatment-resistant or refractory IgAV cases of all ages [2]. It did successfully induce remission in this patient. Thus far, no case reports were published on the use sirolimus in adult-onset IgAV. Sirolimus as a maintenance therapy was chosen based on its therapeutic mechanism as an mTOR inhibitor and its documented use in transplant recipients with recurrent IgA nephropathy [19].
Conclusions
We report on our experience with treating severe systemic refractory IgAV with intensive immunosuppression. Aggressive treatments were warranted to induce and maintain remission in our patients with a high burden of disease. The lack of treatment guidelines gives additional challenges in navigating treatment decisions and predicting treatment outcome and prognosis. A measured and considered approach is required for balancing disease suppression and treatment related complications.
Further studies on the role of biologics and other novel agents in severe IgAV will be highly valuable. The earlier use of biologics could potentially mitigate the burden of treatment-related toxicity. The challenges of balancing the need for immunosuppression and the resulting treatment-related toxicity will not be lost on astute clinicians. Ultimately, the infective complications combined with other significant comorbidities led to shortened survival.
Figures
Tables
Table 1.. IgAV cases.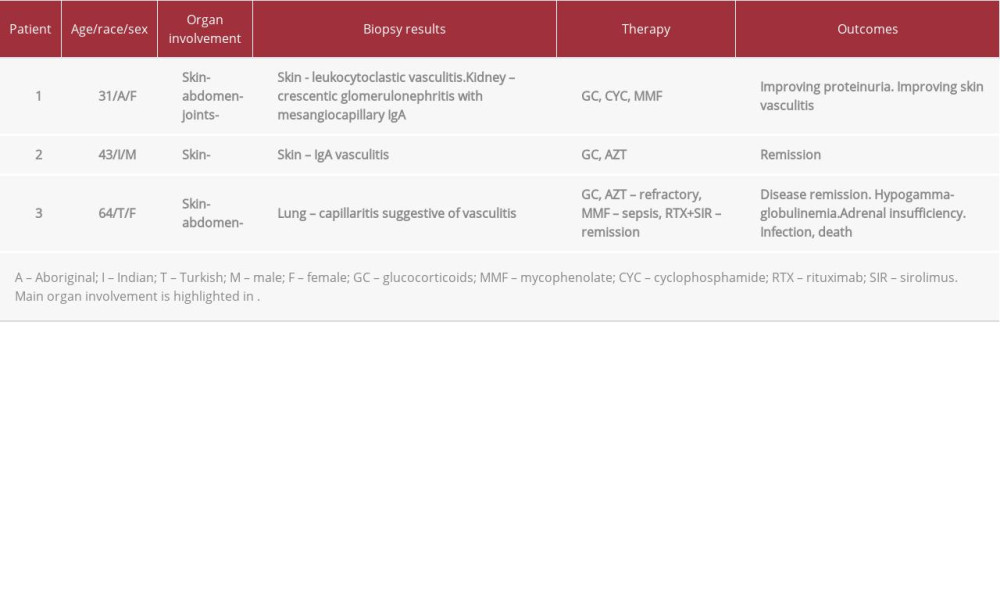
References:
1.. Hocevar A, Tomsic M, Jurcic V, Predicting gastrointestinal and renal involvement in adult IgA vasculitis: Arthritis Res Ther, 2019; 21(1); 302
2.. Hernandez-Rodriguez J, Carbonell C, Miron-Canelo JA, Rituximab treatment for IgA vasculitis: A systematic review: Autoimmun Rev, 2020; 19(4); 102490
3.. Tracy A, Subramanian A, Adderley NJ, Cardiovascular, thromboembolic and renal outcomes in IgA vasculitis (Henoch-Schonlein purpura): A retrospective cohort study using routinely collected primary care data: Ann Rheum Dis, 2019; 78(2); 261-69
4.. Hocevar A, Tomsic M, Rotar Z, IgA vasculitis in adults: Few certainties and many uncertainties: Ann Rheum Dis, 2020; 79(4); e47
5.. Suzuki H, Yasutake J, Makita Y, IgA nephropathy and IgA vasculitis with nephritis have a shared feature involving galactose-deficient IgA1-oriented pathogenesis: Kidney Int, 2018; 93(3); 700-5
6.. Pertuiset E, Liote F, Launay-Russ E, Adult Henoch-Schonlein purpura associated with malignancy: Semin Arthritis Rheum, 2000; 29(6); 360-67
7.. Lopez-Mejias R, Castaneda S, Genre F, Genetics of immunoglobulin-A vasculitis (Henoch-Schonlein purpura): An updated review: Autoimmun Rev, 2018; 17(3); 301-15
8.. Audemard-Verger A, Pillebout E, Guillevin L, IgA vasculitis (Henoch-Shonlein purpura) in adults: Diagnostic and therapeutic aspects: Autoimmun Rev, 2015; 14(7); 579-85
9.. Maritati F, Canzian A, Fenaroli P, Vaglio A, Adult-onset IgA vasculitis (Henoch-Schonlein): Update on therapy: Presse Med, 2020; 49(3); 104035
10.. Nossent J, Raymond W, Keen HI, Morbidity and mortality in adult-onset IgA vasculitis: A long-term population-based cohort study: Rheumatology (Oxford), 2021 [Online ahead of print]
11.. Walters GD, Willis NS, Craig JC, Interventions for renal vasculitis in adults. A systematic review: BMC Nephrol, 2010; 11; 12
12.. Ozen S, Marks SD, Brogan P, European consensus-based recommendations for diagnosis and treatment of immunoglobulin A vasculitis-the SHARE initiative: Rheumatology (Oxford), 2019; 58(9); 1607-16
13.. Radhakrishnan J, Cattran DC, The KDIGO practice guideline on glomerulonephritis: Reading between the (guide)lines – application to the individual patient: Kidney Int, 2012; 82(8); 840-56
14.. Al Harash A, Saeli S, Lucke M, Arora S, IgA vasculitis nephritis: A case series and comparison of treatment guidelines: Case Rep Rheumatol, 2020; 2020; 8863858
15.. Lafayette RA, Canetta PA, Rovin BH, A randomized, controlled trial of rituximab in IgA nephropathy with proteinuria and renal dysfunction: J Am Soc Nephrol, 2017; 28(4); 1306-13
16.. Oshikata C, Tsurikisawa N, Takigawa M, An adult patient with Henoch-Schonlein purpura and non-occlusive mesenteric ischemia: BMC Res Notes, 2013; 6; 26
17.. Ronkainen J, Koskimies O, Ala-Houhala M, Early prednisone therapy in Henoch-Schonlein purpura: A randomized, double-blind, placebo-controlled trial: J Pediatr, 2006; 149(2); 241-47
18.. Sohagia AB, Gunturu SG, Tong TR, Hertan HI, Henoch-schonlein purpura – a case report and review of the literature: Gastroenterol Res Pract, 2010; 2010; 597648
19.. Moroni G, Belingheri M, Frontini G, Immunoglobulin A nephropathy. Recurrence after renal transplantation: Front Immunol, 2019; 10; 1332
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133