16 January 2021: Articles 
Pancreatic Gastrinoma, Gastrointestinal Stromal Tumor (GIST), Pheochromocytoma, and Hürthle Cell Neoplasm in a Patient with Neurofibromatosis Type 1: A Case Report and Literature Review
Rare coexistence of disease or pathology
Arif A. Arif1ADEF, Peter T.W. Kim2ABDE, Adrienne Melck2ABDE, Andrew Churg3ABDE, Zachary Schwartz4ABDE, Heather C. Stuart2ABDEFG*DOI: 10.12659/AJCR.927761
Am J Case Rep 2021; 22:e927761






Abstract
BACKGROUND: Neurofibromatosis type 1 (NF1) is a multi-tumor syndrome in which affected patients develop malignancies that are rare in the overall population, such as tumors of neural or endocrine origin.
CASE REPORT: A 67-year-old woman with a clinical diagnosis of NF1 presented with abdominal pain and pneumoperitoneum. She underwent small-bowel resections for a perforated jejunal lesion and a second lesion in the ileum; pathology showed a neurofibroma at the site of the perforation and a 1-cm low-grade GIST, respectively. Additional staging with cross-sectional imaging identified a 3.7-cm pancreatic head mass and a 1.7-cm left adrenal mass; biochemical studies revealed elevated serum gastrin and urinary free metanephrines and catecholamines consistent with pheochromocytoma. Initial surgical management was a left posterior retroperitoneoscopic adrenalectomy. Postoperatively, gallium-68-DOTATOC PET/CT showed uptake in the pancreatic head and a 28-mm left thyroid nodule. Months later, she had an open pancreaticoduodenectomy. Pathology showed pheochromocytoma and a low-grade (G1) gastrinoma involving 2/8 peripancreatic lymph nodes (pT3pN1M0), respectively. Fine-needle aspiration biopsy of the thyroid nodule showed features consistent with a Hürthle cell neoplasm. Genetic testing identified a pathogenic mutation in NF1 and no mutations in BRCA1/2, CDC72, MEN1, or PALB2. The patient continues surveillance, with no evidence of recurrent disease.
CONCLUSIONS: We report the fifth case of gastrinoma associated with NF1 and the first to arise from the pancreas. This case of a pancreatic neuroendocrine tumor was associated with multiple additional neoplasms. Neuroendocrine tumors found in NF1 should raise suspicion of other malignancies.
Keywords: Gastrinoma, Neurofibromatosis 1, Pancreatic Neoplasms, Pheochromocytoma, Adenoma, Oxyphilic, Endocrine Gland Neoplasms, Gastrointestinal Neoplasms, gastrointestinal stromal tumors
Background
Neurofibromatosis type 1 (NF1) is an autosomal-dominant disorder developing from loss of function of the
Case Report
A 67-year-old woman of Asian descent with a clinical diagnosis of neurofibromatosis presented to the Emergency Department with abdominal pain and was found to have pneumoperitoneum on radiograph. She was taken to the operating room on an urgent basis for a suspected diagnosis of perforated viscus. Although not available at the time of initial assessment, the patient had a longstanding history of peptic ulcer disease requiring 3 previous hospital admissions for upper-GI bleeding in which upper endoscopy demonstrated peptic ulcer disease. She was treated for breast cancer (pT2 pN0, ER/PR-positive, HER2-negative) with left mastectomy and sentinel lymph node biopsy in 2011 and had a remote laparotomy for benign ovarian lesion.
Intra-operatively, a small-bowel tumor in the proximal jejunum was identified as the source of the perforation. This was resected, and a hand-sewn end-to-end anastomosis was created just distal to the ligament of Treitz. A second small-bowel tumor was found in the proximal ileum, which was resected locally with primary closure. Perioperatively, a CT of the abdomen and pelvis showed a pancreatic head mass (3.7 cm) and a left adrenal lesion (1.7 cm) (Figure 1). These were not investigated at the initial surgery as priority was given to clinical stabilization of the patient.
The initial post-operative course was unremarkable. The pathology of the lesion causing the perforation was a gastrointestinal neurofibroma and the ileal lesion, a 1-cm GIST with no mitotic figures (Figure 2A). On post-operative day 10, the patient developed an upper-GI bleed with hemodynamic instability.
Angiography identified a bleeding vessel at the jejunal anastomosis but an attempt at endovascular embolization was unsuccessful. The patient was taken to the operating room, where the anastomosis was resected and she was left in discontinuity for 24 h. When she clinically stabilized, she returned to the OR for a duodenojejunostomy and core biopsy of the pancreatic lesion.
The pathology from the pancreas revealed a well-differentiated, grade 1 neuroendocrine tumor with Ki67 index <1%, no mitotic figures, and immunohistochemistry intensely positive for gastrin (Figure 2B). Serum gastrin was elevated at 9045 ng/mL (normal <115) and chromogranin A was 5470 ng/L (normal <94).
Post operatively, she was noted to be hypertensive, with systolic pressures reaching 200 mmHg, which prompted additional biochemical work-up. Twenty-four-hour urine studies showed elevated metanephrines (4.62 umol/d, normal 0.26–1.73) and catecholamines (epinephrine 507 nmol/d, normal <160; norepinephrine 655 nmol/d, normal 89–470).
With a working diagnosis of a gastrinoma in the head of the pancreas and a left adrenal pheochromocytoma, staging was performed. An I123-MIBG scan was performed, which showed avidity in the left adrenal nodule (Figure 3A). After review at a multidisciplinary tumor board meeting about the sequence of treatment, adrenalectomy was recommended as the initial surgery to minimize the risk of perioperative complications during pancreas surgery. The patient was started preoperatively on alpha blockade with doxasozin and underwent an uncomplicated left adrenalectomy using a posterior retroperitonoscopic approach. The pathology showed an R0 resection of a pheochromocytoma: polygonal cells arranged in a nested pattern with increased mitotic rate (>3/10 high-power field) and atypical mitoses (Figure 2C, annotated). Several higher-risk features, including capsular invasion and invasion of peri-adrenal adi-pose tissue, were identified (Figure 2D). Postoperatively, urine catecholamines and blood pressure normalized.
To assess for a distant neuroendocrine tumor, a 68Ga-DOTATOC PET/CT was performed, which showed uptake only in the pancreatic head mass and a left thyroid nodule (Figure 3B). Several months later, she underwent a pancreaticoduodenectomy with portal vein resection and reconstruction. The pathology was consistent with the initial biopsy showing a low-grade pancreatic neuroendocrine tumor with Ki67 <1% and no mitotic figures. Margins were negative and metastases were identified in 2/8 peripancreatic lymph nodes, pT3pN1M0. Incidentally, a 5-mm duodenal low-grade GIST was identified in the resection specimen. Blood gastrin levels returned to normal within 1 month after surgery (59 ng/mL).
Thyroid ultrasound demonstrated a multinodular gland with a hypoechoic nodule in the left mid-thyroid (2.4×3.8×3.9 cm) with a lobulated margin and no echogenic foci (TIRADS 4). There was no cervical lymphadenopathy. A biopsy showed follicular aggregates of oncocytic thyroid epithelial cells with nuclear atypia and scanty colloid consistent with a Hürthle cell neoplasm (Bethesda IV). Serum CEA and calcitonin were normal at 0.9 ug/L and <1 ng/L (normal <5 and <7, respectively), as were serum calcium and intact PTH. Thyroid nodule genetic sequencing was not available at our institution. The patient underwent a left thyroid lobectomy, which showed a 3.9-cm Hürthle cell adenoma with 1 benign perithyroidal lymph node.
The patient underwent formal genetic testing, showing a pathogenic mutation in the NF1 gene and no mutations in BRCA1/2, CDC72, MEN1, or PALB2. She is under surveillance with cross-sectional imaging and serum and urine biochemistry, with no evidence of recurrent disease at the last follow-up appointment.
She continues to take pantoprazole daily for reflux following pancreaticoduodenectomy.
Discussion
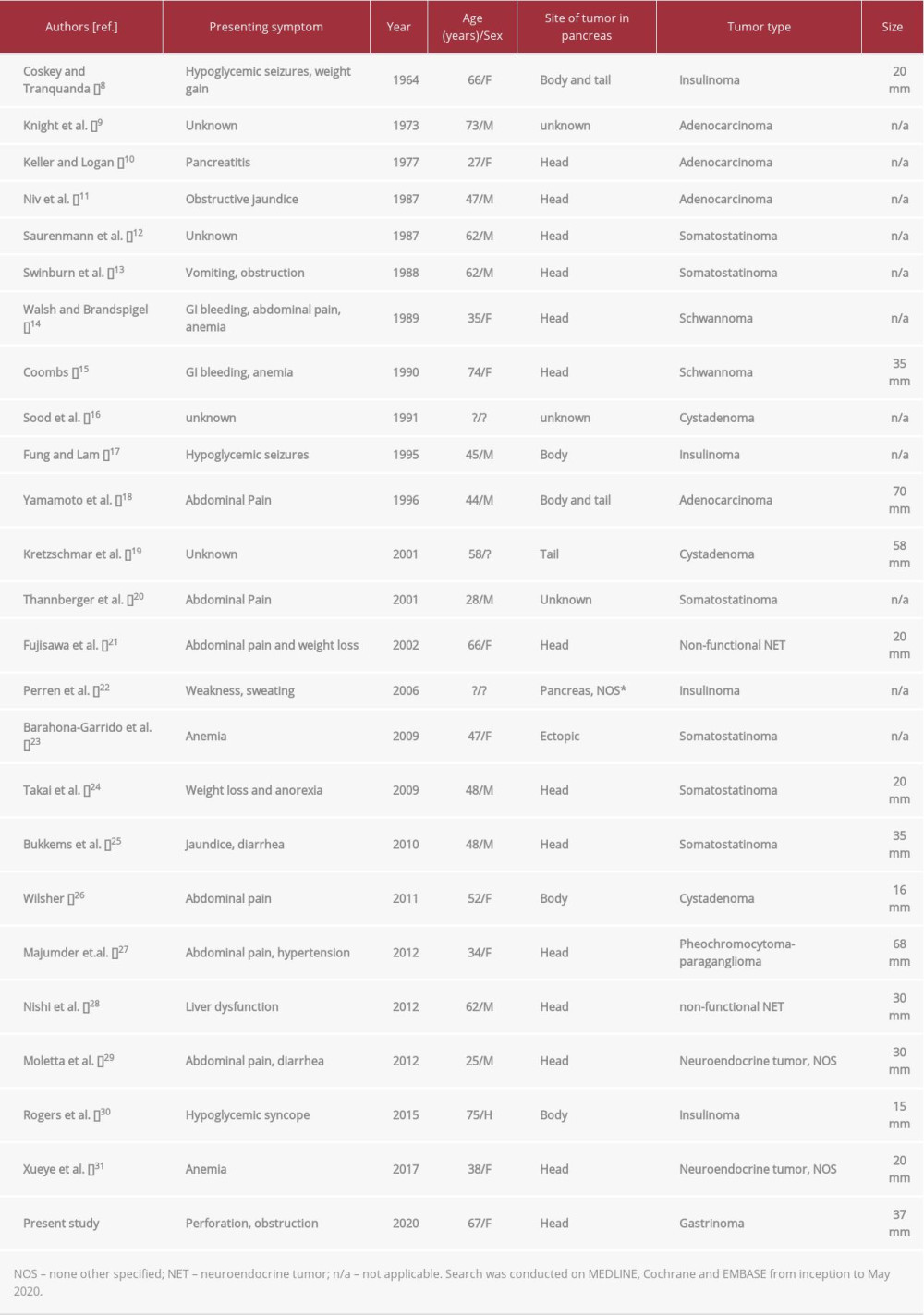
Patients with NF1 develop malignancies that are rare in the overall population, with a propensity for tumors of neural or endocrine origin [2]. Pancreatic neuroendocrine tumors occur at a rate of 0.3 per 100 000 per year [5] and make up 2% of all pancreatic neoplasms in large US-based studies [6]. Of these, 10% were functional neuroendocrine tumors [6], with a prevalence order of: insulinoma> gastrinoma> VIPoma> glucagonoma> somatostatinoma> other hormones [7]. NF1 is not a well-established cause of pancreatic neoplasms; thus, we conducted a literature review to better characterize the reported cases in the context of our patient with a pancreatic neuroendocrine tumor.
A review of all reports of pancreatic neoplasms in NF1 across MEDLINE, Cochrane, and EMBASE from inception to May 2020 identified 24 cases (Table 1). Including the present study, 60% (15/25) of pancreatic neoplasms were neuroendocrine tumors, of which 85% were functional (11/13 with reported function).
Within the functional pancreatic neuroendocrine tumors, 6 cases were reported to be somatostatinomas (54%), 4 insulinomas (36%), and our sole case of a gastrinoma. Our review indicates that the pattern of pancreatic malignancies seen in patients with NF1 differs from that of sporadic pancreatic malignancies. There is an increased proportion of pancreatic neuroendocrine tumors seen in NF1 patients, consistent with a reported propensity for neuroendocrine tumors in NF1 [4], and a propensity for functional neoplasms (85% in NF1 compared to 10% in sporadic) [6]. Loss of heterozygosity in the
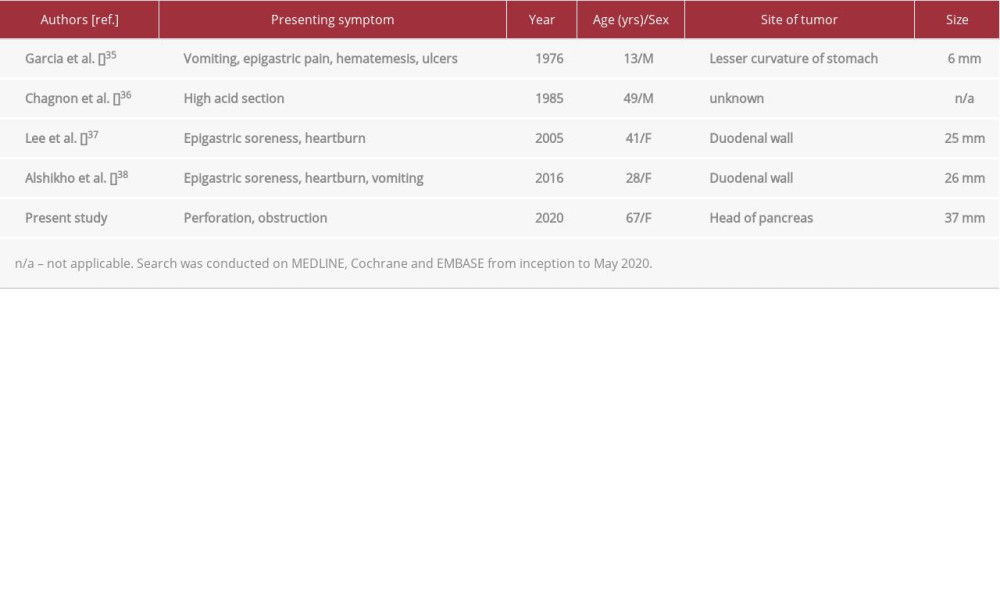
The predominance of functional neuroendocrine tumors in NF1 is well established, with somatostatinomas being the most common [4]. In many cases, the presenting symptom of these tumors is related to a mass effect on surrounding tissue as opposed to the classic functional syndrome of diabetes, diarrhea, and cholelithiasis [13,25,33]. In this case report, the patient presented with a bowel perforation thought to be secondary to a non-functional gastrointestinal neurofibroma. Her functional syndrome was not appreciated until after her initial surgical management; however, in retrospect, her clinical history was characteristic of a Zollinger-Ellison syndrome (ZES). Bowel perforations of the jejunum account for approximately one-half of the presenting complaints in ZES patients [34], making this a likely contributing factor in this case. Unlike the more common somatostatinomas, prior reported cases of gastrinoma in patients with NF1 all had symptoms related to a functional tumor (Table 2).
This is the fifth reported case of gastrinoma in NF1 and the first to be reported in the pancreas. One-half of the gastrinomas with known location were in the duodenum, consistent with the higher incidence of neuroendocrine tumors at this location in NF1; this differs from sporadic gastrinomas, which are more likely to occur in the pancreatic head [39].
Two GISTs were identified in this patient, measuring 10 mm and 5 mm. The incidence of GISTs is known to be increased in NF1, where they often occur as multiple, low-grade tumors in the small bowel [2]. More recently, an association between development of neuroendocrine tumors with GISTs has been uniquely identified in NF1 [4,23,33,40].
An adrenal mass was also identified in this patient and was determined to be a pheochromocytoma after investigation. The characteristics and epidemiology of pheochromocytoma in NF1 has been reviewed elsewhere [2]. Roughly 0.1–5.7% of NF1 patients develop pheochromocytoma with largely solitary and unilateral (84%) tumors [3]. A review of patients with neuroendocrine tumors and NF1 found pheochromocytoma in 6/27 (22%), suggesting a possible link [41]. The co-existence of all 3 neoplasms (neuroendocrine tumor, pheochromocytoma, GIST) in NF1, such as in this report, is exceedingly rare, but has been reported previously [42]. Whether there is an underlying association or common mechanism between these malignancies is unknown; however, discovery of a neuroendocrine tumor in a patient with NF1 should prompt investigation of other potential neoplasms.
One possible explanation for these multiple malignancies is the coincidence of 2 or more genetic syndromes. Multiple endocrine neoplasia syndromes (MEN) predispose individuals to develop tumors in 2 or more endocrine organs. Specifically, multiple endocrine neoplasia 1 (MEN1) has a predilection for developing pancreatic gastrinomas [43]. The present patient had a history of a stage 2 breast cancer that was treated with mastectomy. The combination of these rare genetic syndromes has been reported with a dual
Finally, the presence of thyroid pathology in NF1 has been linked to autoimmune conditions, including alopecia, vitiligo, and autoimmune thyroiditis [46]. There is no known association of NF1 with thyroid malignancy, so the finding of Hürthle cells in this patient could be related to an extrinsic disorder such as autoimmune thyroiditis [47].
Conclusions
This report presents the fifth reported case of gastrinoma associated with NF1 and the first to arise from the pancreas. Neuroendocrine tumors are the most common type of pancreatic malignancy in NF1, and, as in this report, patients can have multiple concurrent malignancies of the gastrointestinal tract and endocrine organs. The identification of a NET in a patient with NF1 should raise the index of suspicion for presence of other malignancies, especially in the context of persistent symptoms.
Figures
References:
1.. Theos A, Korf BR, Pathophysiology of neurofibromatosis type 1: Ann Intern Med, 2006; 144(11); 842-49
2.. Patil S, Chamberlain RS, Neoplasms associated with germline and somatic NF1 gene mutations: Oncologist, 2012; 17(1); 101-16
3.. Walther MM, Herring J, Enquist E, von Recklinghausen’s disease and pheochromocytomas: J Urol, 1999; 162(5); 1582-86
4.. Agaimy A, Vassos N, Croner RS, Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): Clinicopathological spectrum with pathogenetic considerations: Int J Clin Exp Pathol, 2012; 5(9); 852-62
5.. Yao JC, Hassan M, Phan A, One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States: J Clin Oncol, 2008; 26(18); 3063-72
6.. Fitzgerald TL, Hickner ZJ, Schmitz M, Changing incidence of pancreatic neoplasms: A 16-year review of statewide tumor registry: Pancreas, 2008; 37(2); 134-38
7.. Halfdanarson TR, Rubin J, Farnell MB, Pancreatic endocrine neoplasms: Epidemiology and prognosis of pancreatic endocrine tumors: Endocr Relat Cancer, 2008; 15(2); 409-27
8.. Coskey RL, Tranquada RE, Insulinoma and multiple neurofibromatosis: Report of a case: Metabolism, 1964; 13; 312-18
9.. Knight WA, Murphy WK, Gottlieb JA, Neurofibromatosis associated with malignant neurofibromas: Arch Dermatol, 1973; 107(5); 747-50
10.. Keller RT, Logan GM, Adenocarcinoma of the pancreas associated with neurofibromatosis: Cancer, 1977; 39(3); 1264-66
11.. Niv Y, Abu-Avid S, Oren M, Adenocarcinoma of pancreas and duodenum associated with cutaneous neurofibromatosis: Am J Med, 1987; 82(2); 384-85
12.. Saurenmann P, Binswanger R, Maurer R, [Somatostatin-producing endocrine pancreatic tumor in Recklinghausen’s neurofibromatosis. Case report and literature review]: Schweiz Med Wochenschr, 1987; 117(30); 1134-39 [in German]
13.. Swinburn BA, Yeong ML, Lane MR, Neurofibromatosis associated with somatostatinoma: A report of two patients: Clin Endocrinol (Oxf), 1988; 28(4); 353-59
14.. Walsh MM, Brandspigel K, Gastrointestinal bleeding due to pancreatic schwannoma complicating von Recklinghausen’s disease: Gastroenterology, 1989; 97(6); 1550-51
15.. Coombs RJ, Case of the season. Malignant neurogenic tumor of duodenum and pancreas: Semin Roentgenol, 1990; 25(2); 127-29
16.. Sood GK, Chaudhry A, Malhotra V, Pancreatic cystadenoma associated with von Recklinghausen’s disease: Indian J Gastroenterol, 1991; 10(4); 148-49
17.. Fung JW, Lam KS, Neurofibromatosis and insulinoma: Postgrad Med J, 1995; 71(838); 485-86
18.. Yamamoto M, Nakano S, Mugikura M, Pancreatic cancer and hyper-calcemia associated with von Recklinghausen’s disease: J Gastroenterol, 1996; 31(5); 728-31
19.. Kretzschmar M, Ufert S, Hohmann U, [Intraoperative diagnosis of pheochromocytoma preoperative symptoms in a case of Recklinghausen’s disease]: Anaesthesist, 2001; 50(2); 113-17 [in German]
20.. Thannberger P, Wilhelm JM, Derragui A, [Von Recklinghausen’s disease associated with pancreatic somatostatinoma.]: Presse Med Nov 24, 2001; 30(35); 1741-3
21.. Fujisawa T, Osuga T, Maeda M, Malignant endocrine tumor of the pancreas associated with von Recklinghausen’s disease]: J Gastroenterol, 2002; 37(1); 59-67
22.. Perren A, Wiesli P, Schmid S, Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: Molecular analysis of a malignant insulinoma in a NF-1 patient: Am J Surg Pathol, 2006; 30(8); 1047-51
23.. Barahona-Garrido J, Aguirre-Gutierrez R, Gutierrez-Manjarrez JI, Association of GIST and somatostatinoma in a patient with type-1 neurofibromatosis: Is there a common pathway?: Am J Gastroenterol, 2009; 104(3); 797-99
24.. Takai A, Setoyama T, Miyamoto S, Pancreatic somatostatinoma with von Recklinghausen’s disease: Clin Gastroenterol Hepatol, 2009; 7(5); A28
25.. Bukkems SF, Stoot JH, Driessen A, A rare cause of obstructive jaundice and weight loss in Von Recklinghausen’s disease: Neth J Med, 2010; 68(12); 414-17
26.. Wilsher MJ, Metachronous malignant composite phaeochromocytoma and pancreatic mucinous cystadenoma in a patient with neurofibromatosis type 1: Pathology, 2011; 43(2); 170-74
27.. Majumder S, Grabska J, Trikudanathan G, Functional ‘composite’ pheochromocytoma-ganglioneuroma presenting as a pancreatic mass: Pancreatology, 2012; 12(3); 211-14
28.. Nishi T, Kawabata Y, Hari Y, A case of pancreatic neuroendocrine tumor in a patient with neurofibromatosis-1: World J Surg Oncol, 2012; 10; 153
29.. Moletta L MAC, Lico V: Barcelona Spain, 2015
30.. Rogers A, Wang LM, Karavitaki N, Neurofibromatosis type 1 and pancreatic islet cell tumours: An association which should be recognized: QJM, 2015; 108(7); 573-76
31.. Sun X, Yu Y, Jiang Y, Li D, A case of pancreatic neuroendocrine tumor presenting iron deficiency anemia in a patient with neurofibromatosis type 1: Int J Sci, 2017; 6; 51-56
32.. Ruggieri M, Packer RJ, Why do benign astrocytomas become malignant in NF1?: Neurology, 2001; 56(7); 827-29
33.. Thavaraputta S, Graham S, Rivas Mejia AM, Duodenal somatostatinoma presenting as obstructive jaundice with the coexistence of a gastrointestinal stromal tumour in neurofibromatosis type 1: A case with review of the literature: BMJ Case Rep, 2019; 12(1); bcr-2018-226702
34.. Waxman I, Gardner JD, Jensen RT, Peptic ulcer perforation as the presentation of Zollinger-Ellison syndrome: Dig Dis Sci, 1991; 36(1); 19-24
35.. Garcia JC, Carney JA, Stickler GB, Zollinger-Ellison syndrome and neurofibromatosis in a 13-year-old boy: J Pediatr, 1978; 93(6); 982-84
36.. Chagnon JP, Barge J, Henin D, [Recklinghausen’s disease with digestive localizations associated with gastric acid hypersecretion suggesting Zollinger-Ellison syndrome]: Gastroenterol Clin Biol, 1985; 9(1); 65-69 [in French]
37.. Lee WS, Koh YS, Kim JC, Zollinger-Ellison syndrome associated with neurofibromatosis type 1: A case report: BMC Cancer, 2005; 5; 85
38.. Alshikho MJ, Noureldine SI, Talas JM, Zollinger-Ellison syndrome associated with von Recklinghausen disease: Case report and literature review: Am J Case Rep, 2016; 17; 398-405
39.. Donow C, Pipeleers-Marichal M, Schroder S, Surgical pathology of gastrinoma. Site, size, multicentricity, association with multiple endocrine neoplasia type 1, and malignancy: Cancer, 1991; 68(6); 1329-34
40.. Yamamoto R, Kato S, Maru T, The coexistence of somatostatinoma and gastrointestinal stromal tumor in the duodenum of a patient with von Recklinghausen’s disease: Intern Med, 2016; 55(6); 617-22
41.. Griffiths DF, Williams GT, Williams ED, Duodenal carcinoid tumours, phaeochromocytoma and neurofibromatosis: Islet cell tumour, phaeochromocytoma and the von Hippel-Lindau complex: Two distinctive neuroendocrine syndromes: Q J Med, 1987; 64(245); 769-82
42.. Poredska K, Kunovsky L, Prochazka V, Triple malignancy (NET, GIST and pheochromocytoma) as a first manifestation of neurofibromatosis type-1 in an adult patient: Diagn Pathol, 2019; 14(1); 77
43.. Jensen RT, Berna MJ, Bingham DB, Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies: Cancer, 2008; 113(7 Suppl.); 1807-43
44.. Ercolino T, Lai R, Giache V, Patient affected by neurofibromatosis type 1 and thyroid C-cell hyperplasia harboring pathogenic germ-line mutations in both NF1 and RET genes: Gene, 2014; 536(2); 332-35
45.. Jeon YW, Kim RM, Lim ST, Early-onset breast cancer in a family with neurofibromatosis type 1 associated with a germline mutation in BRCA1: J Breast Cancer, 2015; 18(1); 97-100
46.. Nanda A, Autoimmune diseases associated with neurofibromatosis type 1: Pediatr Dermatol, 2008; 25(3); 392-93
47.. Cannon J, The significance of hurthle cells in thyroid disease: Cncologist, 2011; 16(10); 1380-87
Figures
In Press
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952658
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953243
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.952989
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953049
Most Viewed Current Articles
07 Dec 2021 : Case report
22,697,854
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,914
174,914
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
120,026
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,962
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133