16 August 2022: Articles 
Histiocytic Disorder Mimicking a Brain Tumor: A Report of 2 Rare Cases
Challenging differential diagnosis, Diagnostic / therapeutic accidents, Management of emergency care, Rare disease
Frances Xiuyan Feng12BCEF, Negin FarsiDOI: 10.12659/AJCR.935885
Am J Case Rep 2022; 23:e935885






Abstract
BACKGROUND: Histiocytic disorders, a group of disorders with heterogeneous pathogenesis, morphology, and clinical presentation, include Rosai-Dorfman disease, Langerhans cell histiocytosis, and Erdheim-Chester disease. They can mimic primary or metastatic tumors, both clinically and radiologically, when involving the brain. Therefore, it is crucial to present and discuss cases of histiocytic disorder involving the central nervous system (CNS) to provide new information on disease presentation and diagnosis more. In this paper, we present 2 cases of histiocytic lesions involving the brain and mimicking primary brain tumors.
CASE REPORT: Case 1: A 65-year-old man presented with increasing memory loss, confusion, and depression. CT scans showed an isolated 2.9×2.0×0.6 cm intracranial hypothalamic lesion. Case 2: A 61-year-old woman presented with dizziness and confusion for 3 weeks and headaches for 1 day. MRI showed a single 5.0×4.0×3.3 cm extra-axial, dural-based, avidly enhancing, well-defined lesion along the left parietal convexity causing mass effect upon the underlying brain parenchyma, left atrial effacement, and minimal vasogenic edema.
CONCLUSIONS: Histiocytic disorders are relatively rare in the CNS compared with other locations and mimic more common entities in the brain, such as glioma or metastatic tumors. Despite its rarity, one should remain aware of the condition and consider it in the differential diagnosis. This article provides a brief review and adds pivotal data to the literature.
Keywords: Histiocytic Disorders, Malignant, Histiocytosis, Langerhans-Cell, Histiocytosis, Sinus, Aged, Brain Neoplasms, Diagnosis, Differential, Female, Humans, Magnetic Resonance Imaging, Male, Middle Aged
Background
Langerhans cell histiocytosis (LCH) is a clonal neoplastic proliferation of Langerhans cells typically presenting in childhood with a male predilection. LCH can be localized to a single site, can occur in multiple sites of a single system or can be more disseminated and multisystem. The WHO classification of LCH includes NOS, monostotic, polyostotic, and disseminated. The predominant sites of involvement in the solitary form are bone and adjacent soft tissue. Patients with the unifocal disease are usually older children or adults who most commonly present with a lytic bone lesion; solitary lesions at other sites present as mass lesions or enlarged lymph nodes. Patients with uni-system multifocal disease are usually young children presenting with multiple or sequential destructive bone lesions, often associated with adjacent soft-tissue masses. Patients with multisystem involvement are infants who present with fever, cytopenia, skin and bone lesions, and hepatosplenomegaly. The clinical course of LCH is related to the staging of the disease at presentation, with 99% survival for unifocal diseases and 66% mortality for young children with multisystem involvement who do not respond promptly to therapy. CNS involvement is rare, and isolated intracranial disease in adults is rarely reported [1–3].
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare, idiopathic, nonneoplastic disorder that usually presents with massive painless lymphadenopathy, fever, leukocytosis, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Most cases of RDD occur during the first and second decades of life. The most common and prominent site of involvement is the cervical lymph nodes. Patients with localized nodal disease usually have a chronic, indolent course. Extranidal involvement of the liver, kidney, and lower respiratory tract, as well as signs of overt immune dysfunction are associated with poor prognosis. RDD rarely involves the CNS. The isolated intracranial disease is sporadic and can be mistaken for other pathologies in imaging studies [4].
Herein, we present a case of intracranial dural-based LCH initially considered a meningioma and a case of isolated RDD suspicious for glioblastoma.
Case Report
CASE 1:
A 65-year-old man presented with increasing memory loss, confusion, and depression. CT scans showed an isolated intracranial hypothalamic lesion. On magnetic resonance imagining (MRI), the lesion presented as a well-defined, inhomogeneous, enhancing mass involving the optic chiasm (Figure 1A, 1B) and suspicious for glioblastoma. The patient underwent surgical resection.
The specimen consisted of light brown soft-tissue fragments that measured 2.9×2.0×0.6 cm. On microscopic examination, abundant large histiocytes admixed with lymphocytes, plasma cells, and mature eosinophils were observed in a fibrous background. No necrosis or normal brain parenchyma was identified. The histiocytes revealed abundant pale eosinophilic cytoplasm and oval nuclei with irregular nuclear membranes, prominent nuclear grooves, fine chromatin, and indistinct nucleoli (Figure 2A–2D). The histocytes were positive for CD1a, CD68, S100, and focally positive for Langerin (Figure 2E–2H). Morphologic and immunohistochemical features were diagnostic of LCH. The patient subsequently died due to complications of hypothyroidism, adrenal insufficiency, and post-surgical diabetes insipidus.
CASE 2:
A 61-year-old woman presented with dizziness and confusion for 3 weeks and headaches for 1 day. MRI showed a single 5.0×4.0×3.3 cm extra-axial, dural-based, avidly enhancing, well-defined lesion along the left parietal convexity causing a mass effect on the underlying brain parenchyma, left atrial effacement, and minimal vasogenic edema (Figure 3A, 3B). Meningioma was initially considered. After a complete work-up, the mass was resected.
The surgical specimen consisted of pink-tan tissue fragments measuring 0.9×0.7×0.3 cm and 5.3×3.8×0.3 cm, respectively. As shown in Figure 4A–4D, the H&E-stained slides demonstrated brain parenchyma diffusely infiltrated by sheets of large histiocytes with prominent plasma cells and mature lymphocytes. The histiocytes revealed abundant clear, eosinophilic cytoplasm with conspicuous intracytoplasmic, and engulfed lymphocytes (emperipolesis). Some of the plasma cells contained Russell bodies. The nuclei of histocytes were large, round, and vesicular with delicate nuclear membranes and prominent nucleoli.
Immunohistochemically, the histiocytes were reactive to CD68 and S100 but negative for CD1a (Figure 4E–4G). In-situ hybridization (ISH) stains demonstrated polyclonal CD138-positive plasma cells (Figure 4H–4J). The morphological and immunohistochemical profiles and ISH studies were diagnostic of RDD.
Discussion
Based on the most recent classification of histiocytic lesions, which consist of 5 groups of diseases (L, C, R, M, and H) [5], LCH belongs to the L group, which also includes intermediate cell histiocytosis (ICH), Erdheim-Chester disease (ECD), and mixed LCH/ECD, while RDD is classified as “C” or “R” group depending on the location, as shown in Table 1.
About 4–25% of LCH cases involve the CNS clinically. Cases most frequently present with pituitary dysfunction, including diabetes insipidus and mass effect secondary to intra-axial or extra-axial lesions. Radiologists describe the CNS findings as tumorous/granulomatous lesions or non-tumorous/non-granulomatous lesions (neurodegenerative) or atrophic [6]. However, considerable overlap between these subgroups and findings of all 3 subgroups can occur in a single patient.
Intracranially, the characteristic radiologic lesion involves the hypothalamic-pituitary axis with an enhancing suprasellar mass and thickening of the infundibular stalk. Loss of the standard hyperintense signal on T1-weighted images from the posterior pituitary may also be detected. Calvarial bone lesions show preferential involvement of the outer table compared to the inner table, thus producing a classic “bone in bone” appearance. An associated scalp or soft-tissue component is often seen [7]. In our case, the isolated intracranial LCH occurred as is most commonly observed in the hypothalamic area, although the patient presented with confusion without pituitary symptoms. However, the patient reported having mild memory loss for a few months with sudden exacerbation over a few days before presenting to the Emergency Department. In addition, the patient reportedly had a recent fall requiring a cane to enable balance. Diabetes insipidus was not present before the craniotomy. Tan et al reported a similar case that occurred in the seller area in a 50-year-old woman. Their patient presented with DM-like symptoms, polydipsia, and polyuria, for over 3 months [8].
Microscopically, the LCH cells are oval with grooved, folded, indented, or lobed nuclei with fine chromatin, inconspicuous nucleoli, and thin nuclear membranes. The cytoplasm is moderately abundant and slightly eosinophilic. A variable number of eosinophils, histocytes, neutrophils, and small lymphocytes are present. Plasma cells are sparse. The classic morphologic features were present in our case. In the early stage, LCH cells predominate, along with eosinophils and neutrophils, while in the late stages of the disease, the LCH cells are decreased, with increased foamy macrophages and fibrosis. Tumor cells consistently express CD1a, langerin, and S100 protein. Crucial immunohistochemical stains differentiate LCH from other histiocytic lesions, including CD1a, Langerin, S100, CD68, HLA-DR, and vimentin. The ultrastructural hallmark of LCH is the cytoplasmic tennis-racket-shaped Birbeck granules.
LCH should be differentiated from Erdheim-Chester disease. The latter is considered another clonal systemic histiocytic proliferative disease involving any organ and tissue belonging to the “L” group, just like LCH. The clinical and radiological features can be very similar between the 2 diseases. However, skeletal involvement occurs in 95% of ECD cases, and there is typically bilateral and symmetric involvement of the long bones, which is much less common in LCH. CNS involvement occurs in 20–30% of patients with ECD and produces variable symptoms depending on tumor location [9]. The most severe neurological complication is a neurodegenerative cerebellar disease, present in 15–20% of patients with ECD. CNS involvement is a significant prognostic factor, constituting an independent predictor of death. Morphologically, the lesional histocytes have single small nuclei and foamy or compact eosinophilic cytoplasm. Touton cells with a central ring of nuclei are frequently observed. Fibrosis is present in most cases and is sometimes abundant. Reactive small lymphocytes, plasma cells, and neutrophils are frequently present. The infiltration is easily misdiagnosed as a reactive process. The ECD histocytes can express the typical macrophage markers– CD14, CD68, and CD163 – but lack the Langerhans cell markers CD1a, S100, and langerin. Notably, up to 20% of patients with ECD also have Langerhans cell histiocytosis [5], and BRAF V600E mutation is reported in 50% of both LCD and ECD [10].
Therefore, immunohistochemical stains should be used to distinguish these entities instead of BRAF V600E mutation. Careful evaluation of our case did not reveal an ECD component. Another critical issue is that histopathology and phenotype are not distinct between ECD and extracutaneous or disseminated juvenile xanthogranuloma. When suspected ECD patients do not present with typical bilateral and symmetric involvement of the long bones, molecular testing should be performed.
Most cases of Rosai-Dorfman disease occur during the first and second decade of life and typically manifest as cervical lymph node enlargement, but other peripheral or central lymph node groups can be affected. In over 25% of the cases, RDD involves extranodal sites, including the CNS. Cerebral involvement usually manifests as dural-based lesions, often clinically and radiologically mistaken for meningiomas, just like this case. Intraparenchymal involvement is far less common, typically mimicking lymphoma or intraparenchymal tuberculous granulomas. Furthermore, intraventricular lesions have been reported in some cases. Involvement of the intramedullary spinal cord, cavernous sinus, and retroocular region of the orbit is infrequent. No neurodegenerative pattern has been described in patients with RDD, unlike LCH and ECD [11–14]. Histologically, the lesions consist of numerous characteristic large histiocytes enmeshed in a variably cellular mixed inflammatory infiltrate composed of plasma cells often containing Russell bodies, lymphocytes, neutrophils, foamy macrophages, and rare eosinophils. The large histiocytes contain abundant eosinophilic cytoplasm and demonstrate emperipolesis with intracytoplasmic lymphocytes, plasma cells, or neutrophils. The nuclei of histocytes range from round or oval to kidney bean-shaped with fine or vesicular chromatin and prominent nucleoli, which can be large. The histiocytes are variable in number and distribution. Microabscesses may be present. The large histiocytes express S100, CD68, and CD163 and are negative for CD1a, as shown in the present case. Molecular studies on involved tissue fail to demonstrate evidence of clonality, in keeping with their presumed reactive nature. The comparisons of the pathological features among the most common histiocytic disorders with CNS involvement are shown in Table 2.
Conclusions
LCD, ECD, and RDD are the most common histiocytic lesions with morphologic similarities. They are rarely involved in the CNS and present similar clinical and radiological features with primary CNS tumors. In this report, both cases of histiocytic lesions involving the brain were initially diagnosed as primary brain tumors clinically. Our experiences will help in accurate diagnoses of those entities involving the brain. As mimics of meningioma or glioma, histiocytic lesions should be included in the differential diagnoses due to the similarity of clinical symptoms and radiology features among those entities.
Figures
References:
1.. : WHO Classification of Tumours Editorial Board World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues Resvise, 2017, Lyon, International Agency for Research on Cancer
2.. Duarte-Celada WR, Thakolwiboon S, Brandi L, Adult Langerhans cell histiocytosis of the central nervous system: Proc (Bayl Univ Med Cent), 2020; 33(4); 603-5
3.. Cai S, Zhang S, Liu X, Solitary Langerhans cell histiocytosis of frontal lobe: A case report and literature review: Chin J Cancer Res, 2014; 26(2); 211-14
4.. Boissaud-Cooke MA, Bhatt K, Hilton DA, Isolated Intracranial Rosai-Dorfman disease. Case report and review of the literature: World Neurosurg, 2020; 137; 239-42
5.. Emile JF, Abla O, Fraitag S, Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages: Blood, 2016; 127(22); 2672-81
6.. Grois N, Fahrner B, Arceci RJ, Central nervous system disease in Langerhans cell histiocytosis: J Pediatr, 2010; 156(6); 873-81 e871
7.. Wang Y, Camelo-Piragua S, Abdullah A, Neuroimaging features of CNS histiocytosis syndromes: Clin Imaging, 2020; 60(1); 131-40
8.. Tan H, Yu K, Yu Y, Isolated hypothalamic-pituitary Langerhans’ cell histiocytosis in female adult: A case report: Medicine (Baltimore), 2019; 98(2); e13853
9.. Luna LP, Drier A, Aygun N, MRI features of intra-axial histiocytic brain mass lesions: Clin Radiol, 2021; 76(2); 159.e119-59.e128
10.. : World Health Organization Classification of Tumours of the Central Nervous System, 2021, Lyon, International Agency for Research on Cancer
11.. Petraglia AF, Davick JJ, Mandell JW, Rosai-Dorfman disease: A less common cause of leptomeningeal and nerve root enhancement: Neurohospitalist, 2020; 10(4); 309-13
12.. Varrassi M, Corridore A, Tommasino E, MR imaging of cerebral involvement of Rosai-Dorfman disease: A single-centre experience with review of the literature: Radiol Med, 2021; 126(1); 89-98
13.. Wen JH, Wang C, Jin YY, Radiological and clinical findings of isolated meningeal Rosai-Dorfman disease of the central nervous system: Medicine (Baltimore), 2019; 98(19); e15365
14.. Taufiq M, Khair A, Begum F, Isolated intracranial Rosai-Dorfman disease: Case Rep Neurol Med, 2016; 2016; 1972594
Figures
Tables
 Table 1.. 2016 Revised classification of histiocytosis and neoplasms of the macrophage-dendritic cell lineages.
Table 1.. 2016 Revised classification of histiocytosis and neoplasms of the macrophage-dendritic cell lineages.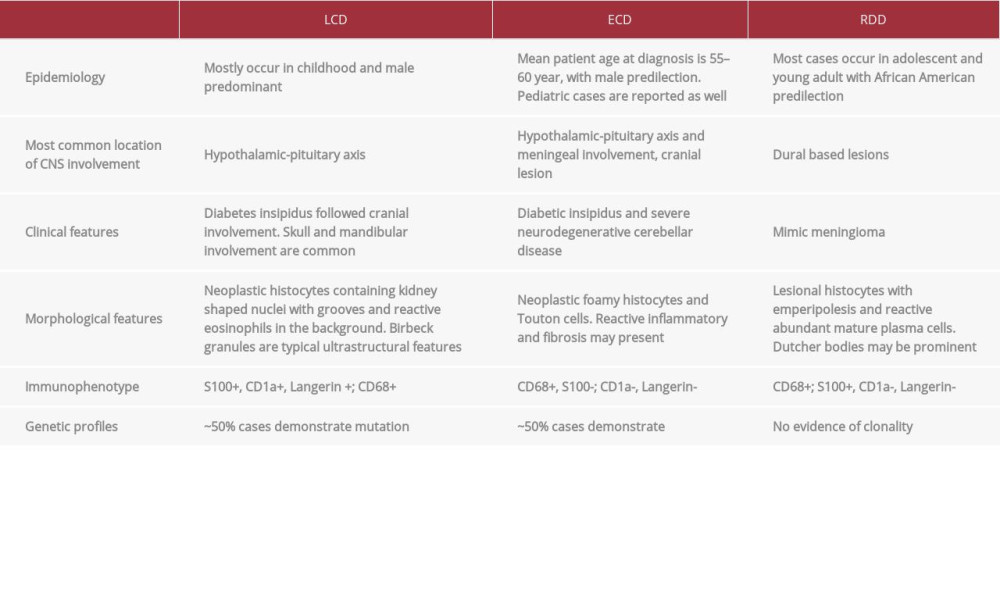 Table 2.. Comparison of CNS involvement of three most common histiocytic lesions.Table 1.. 2016 Revised classification of histiocytosis and neoplasms of the macrophage-dendritic cell lineages.Table 2.. Comparison of CNS involvement of three most common histiocytic lesions.
Table 2.. Comparison of CNS involvement of three most common histiocytic lesions.Table 1.. 2016 Revised classification of histiocytosis and neoplasms of the macrophage-dendritic cell lineages.Table 2.. Comparison of CNS involvement of three most common histiocytic lesions. In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133