30 April 2023: Articles 
Atypical Hemolytic Uremic Syndrome in a Pregnant Patient with a Thrombomodulin Gene Variant Treated with Plasma Exchange and Eculizumab
Challenging differential diagnosis, Unusual setting of medical care, Rare disease
Lakshmi KannanDOI: 10.12659/AJCR.938896
Am J Case Rep 2023; 24:e938896






Abstract
BACKGROUND: Hemolytic uremic syndrome (HUS) includes the triad of thrombocytopenia, microangiopathic hemolytic anemia, and acute renal failure. The atypical form of HUS is a rare disease characterized by complement overactivation, and it can be from genetic or acquired causes. Genetic causes involve mutation in one of the factors in the alternative complement pathway or inhibitors. Malignant hypertension and pregnancy are the most important acquired causes. The optimal management of patients with aHUS is with eculizumab, which is recombinant antibody against human complement component C5.
CASE REPORT: This report describes the case of a 25-year-old woman with frequent hospitalizations for poorly controlled hypertension who presented at 20 weeks of gestation with headache, vomiting, and a blood pressure of 230/126 mmHg. The patient had acute kidney injury with hematuria and proteinuria, and kidney biopsy showed hypertensive arteriolar nephrosclerosis and fibrinoid arteriolar necrosis consistent with thrombotic microangiopathy. Further work-up with a genetic panel showed heterozygosity for the thrombomodulin (THBD) gene. She was started on treatment with plasma exchange and eculizumab, a recombinant monoclonal antibody that inhibits terminal complement activation at the C5 protein. The patient responded well to the treatment at the time of her initial outpatient follow-up.
CONCLUSIONS: This case shows the potential of severe renal manifestation of aHUS, and the need for a kidney biopsy in cases of severe uncontrolled hypertension presenting with kidney injury. If evidence of aHUS is found, prompt treatment with plasma exchange and eculizumab should be initiated.
Keywords: Atypical Hemolytic Uremic Syndrome, Hypertension, Thrombotic Microangiopathies, Female, Pregnancy, Humans, Adult, Plasma Exchange, Thrombomodulin, Purpura, Thrombotic Thrombocytopenic, Acute Kidney Injury
Background
Atypical hemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy (TMA) characterized by microangiopathic hemolytic anemia, thrombocytopenia, and microthrombi, leading to ischemic tissue injury in the renal vasculature. It is associated with fibrinoid necrosis of the arterioles and glomerular capillary tufts [1,2], as well as with dysregulation of alternative complement pathways, which can be caused by genes regulating alternative complement pathways or autoantibodies inhibiting complement-regulating proteins [3]. There are many factors – genetics, diseases, and drugs – that can cause aHUS. Malignant hypertension and pregnancy are 2 important underlying factors.
TMA syndrome can cause damage to the endothelium, which may lead to elevated blood pressure. However, severe hyper-tension can further damage the endothelium and renal vasculature, resulting in fibrinoid necrosis of arterioles and glomerular capillary tufts. Patients with hypertension-associated TMA should receive aggressive blood pressure control and supportive treatment to prevent further kidney injury. A recent case series found that genetic predisposition to using the alternative complement pathway was identified in patients with TMA triggered by severe hypertension [4]. Despite controlled blood pressure, 8 out of 9 patients showed progression to ESRD, indicating dysregulation of the alternative complement pathway as the cause of renal impairment in malignant hypertension [5].
The therapeutic focus for aHUS has been to resolve dysregulation in the complement system. Plasma therapy was previously the first-line treatment option for aHUS, but its efficacy varies depending on the genetic abnormalities involved [6]. Eculizumab, a monoclonal antibody to terminal C5, has been introduced and has shown superior outcomes compared to plasma therapy. Eculizumab is effective in preventing organ damage and premature death in aHUS patients. It has helped in controlling the disease and accelerating related diagnosis and treatment through clinical, functional, and molecular research [7].
This report is of a 25-year-old woman with a history of poorly controlled hypertension who presented at 20 weeks of gestation with hypertension, hypertensive arteriolar nephrosclerosis, and atypical hemolytic uremic syndrome, who was heterozygous for the thrombomodulin (THBD) gene and responded to treatment with plasma infusion and eculizumab.
Case Report
A 25-year-old woman with poorly controlled hypertension presents to our hospital with nausea, vomiting, severe headache, uncontrolled hypertension, and acute kidney injury (AKI) on a background of chronic kidney disease (CKD). She was initially diagnosed with hypertension at Employee Health 3 years before and she has had frequent Emergency Department (ED) visits with severe headaches, and vomiting. Her outpatient antihypertensives were clonidine 0.2 mg twice daily and lisinopril 20 mg daily. She has been very non-compliant with her medications (including not refilling her prescriptions). At that time, her blood work showed sodium 141 mmol/L, potassium 3.9 mmol/L, bicarbonate 27 mmol/L, blood urea nitrogen 8.6 mg/dL, and creatinine 0.7 mg/dL, with estimate glomerular filtration rate (eGFR) >90 ml/min/1.73 m2.
During subsequent visits for uncontrolled hypertension, cloni-dine was switched to chlorthalidone 25 mg daily and amlodipine 5 mg daily. At that time, her kidney function was stable, but her urine analysis showed 30 mg/dL protein. Secondary work-up showed thyroid-stimulating hormone (TSH) 1.1 uIU/ml, serum aldosterone 1.1 ng/dL (normal <15 ng/dL), plasma renin activity 3.19, and aldosterone-to-renin ratio 0.3. Renal Doppler ultrasound showed right kidney size 10.3 cm, left kidney size 10.56 cm, and normal flow parameters (Table 1).
After a year of failing to attend follow-up appointments, the patient presented to the hospital 9-weeks pregnant, with nausea, vomiting, 10/10 headache, and uncontrolled hypertension (207/119 mmHg). The blood work showed sodium 138 mmol/L, potassium 3.4 mmol/L, bicarbonate 27 mmol/L, blood urea nitrogen (BUN) 17 mg/dL, and creatinine of 0.91 mg/dL, with eGFR 83 ml/min/1.73 m2. The complete blood count showed anemia, with hemoglobin 6.7 g/dL (requiring 2 units of packed red blood cell transfusion) and thrombocytopenia with platelet count of 95 000/microliter of blood. She was started on labetalol and diltiazem. She also had to be upgraded to the Intensive Care Unit (ICU) for a brief period on labetalol drip, and the only medications that helped control her BP were labetalol 400 mg every 8 hours, alpha methyldopa 250 mg every 12 hours, and hydralazine 75 mg every 6 hours. Further work-up with peripheral smear showed schistocytes consistent with hemolytic anemia. The ADAMTS 13 level was 12% but the inhibitor was negative at <0.4, and secondary work-up for autoimmune process was negative, as shown in Table 2.
A kidney biopsy was obtained, which showed fibrinoid arteriolar necrosis consistent with thrombotic microangiopathy, perihilar focal segmental glomerulosclerosis, mild interstitial fibrosis, and mild tubular atrophy (Figure 1).
The patient received 3 days of fresh frozen plasma (FFP) for possible hereditary thrombocytopenic purpura but her creati-nine stabilized between 2 and 3 mg/dL and she was discharged home on furosemide 20 mg twice daily in addition to hydralazine 25 mg every 6 hours, labetalol 200 mg every 8 hours, and methyldopa 250 mg every 12 hours.
The patient missed her follow-up appointments and subsequently returned to the hospital 20-weeks pregnant, with headache, vomiting, and BP of 230/126 mmHg. On physical examination, her temperature was 36.5°C, heart rate 90 beats per minute, blood pressure 212/135 mmHg, and oxygen saturation 99% on room air. She received hydralazine 10 mg i.v. twice, with labetalol 400 mg and methyldopa 250 mg, and her BP improved to 168/108 mmHg. She was also given 50 mcg of fentanyl i.v. for severe headache.
Because of uncontrolled HTN and its complications, the decision was initiate labor. During her entire stay, her BP was very labile, requiring esmolol drip, and nicardipine drip during labor, which was switched to amlodipine and losartan after delivery.
She received 2 FFP units on the day of admission and 2 FFP units on hospital day 2. Subsequently, she had worsening shortness of breath, chest pain, and increased oxygen requirements after transfusion due to transfusion-associated circulatory collapse (TACO). She was upgraded to the ICU for worsening respiratory status and was ultimately intubated for respiratory distress. TACO resolved with the initiation of hemodialysis (volume overload in the setting of CKD i.v.) and the switch to plasma exchanges. A complement-mediated TMA panel was sent to the Mayo Clinic, as shown in Table 3.
The patient was found to be heterozygous for the THBD gene (thrombomodulin)-cDNA change-c. 1878G>C, amino acid change-p. Glu560Gn. Eculizumab was started at the time of discharge with the belief that the patient’s kidney function would improve, as the final kidney biopsy only showed mild interstitial fibrosis and tubular atrophy. At the outpatient follow-up visit, she had received 4 doses of eculizumab and her hemoglobin and platelets had stabilized.
Discussion
Thrombotic microangiopathy is a category of acquired and hereditary diseases characterized by microvascular endothelial damage. Microangiopathic hemolytic anemia (schistocytes on the peripheral smear), thrombocytopenia, and end-organ damage are the clinical hallmarks. Conditions that share these features include hemolytic uremic syndrome (HUS), thrombotic thrombocytopenic purpura (TTP), and atypical hemolytic uremic syndrome (aHUS) caused by accelerated or malignant hypertension, autoimmune diseases like systemic lupus erythematosus, antiphospholipid syndrome, HIV, pre-eclampsia/eclampsia, disseminated intravascular coagulation (DIC), drugs like interferon, calcineurin inhibitors, quinine, cocaine, i.v. Opana, heroin, and chemotherapy [8].
Atypical hemolytic uremic syndrome is caused by mutations in genes encoding effector or regulatory elements of the complement or coagulation pathways [9]. In genetically susceptible patients with aHUS, it may occur spontaneously or as a result of malignancy, autoimmune illness, bacteria and viruses, drugs, pregnancy, transplant rejection, or malignant hypertension [10].
In malignant hypertension, aHUS is caused by platelet activation and thrombosis from mechanical stress. It is debatable whether primary HUS is complicated by secondary accelerated hypertension or hypertensive emergency, or whether primary hypertensive emergency leads to secondary HUS. The penetrance of aHUS is incomplete, and in most cases, a “second hit” such as hypertension is needed to manifest the disease [11].
This phenomenon was proven in our patient, where complement is activated by accelerated hypertension and pregnancy in a susceptible young female with the genetic variant THBD and given the lack of intrinsic ability to regulate, the complement pathway spirals out of control.
There are case reports where patients present with severe hypertension but subsequently show features of aHUS [12–14]. Kidney biopsy showed arteriolar TMA in 100 non-elderly patients presenting with severe hypertensin and renal failure in a 2018 report by Larsen et al [15].
The risk of end-stage renal disease and disease recurrence after transplantation are high with aHUS [16]. In a recent study [4] on 9 patients with TMA associated with hypertension, 67% had impaired complement regulation and invariably advanced to end-stage renal disease, and recurrence after transplantation was common.
Historically, aHUS had a poor prognosis, with approximately 50% of patients progressing to end-stage renal disease (ESRD). Plasma infusion or exchange (PE) was the standard of care prior to the introduction of eculizumab, but it did not address the underlying pathology. The efficacy of PE varies depending on the underlying genetic mutations of the patient. A study analyzing clinical outcomes in 273 patients with aHUS treated with PE found that nearly 70% of patients with genetic mutations either died or became dialysis-dependent during the first episode or within 3 years of the first manifestation [16]. PE also has adverse effects, such as hypotension, symptomatic hypocalcemia, allergic reactions, and catheter-related thrombosis, especially in pediatric patients [17]. Eculizumab, a humanized monoclonal antibody, was approved by the FDA in 2011 and has brought about a paradigm shift in the management of aHUS, with clinical trials showing its superiority to PE.
Eculizumab has contributed to improved outcomes and prognosis for patients with aHUS [18].
Conclusions
In clinical practice, most patients who present with severe hypertension and chronic kidney disease are inferred to have arterial nephrosclerosis. However, it is important to screen for TMA in patients who present with severe uncontrolled hyper-tension, AKI, and other features of TMA. Kidney biopsy and further genetic testing, including abnormalities of the alternative complement pathway and secondary factors, can be done after weighing the risks and benefits. Prompt treatment with plasma exchange and terminal complement factor inhibitor is necessary to prevent progression of kidney disease.
References:
1.. Joseph C, Gattineni J, Complement disorders and hemolytic uremic syndrome: Curr Opin Pediatr, 2013; 25; 209-15
2.. Claes KJ, Massart A, Collard L, Belgian consensus statement on the diagnosis and management of patients with atypical hemolytic uremic syndrome: Acta Clin Belg, 2018; 73; 80-89
3.. Campistol JM, Arias M, Ariceta G, An update for atypical haemolytic uraemic syndrome: Diagnosis and treatment. A consensus document: Nefrologia, 2015; 35; 421-47
4.. Timmermans SAMEG, Abdul-Hamid MA, Vanderlocht J, Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities: Kidney Int, 2017; 91; 1420-25
5.. Zhang B, Xing C, Yu X, Renal thrombotic microangiopathies induced by severe hypertension: Hypertens Res, 2008; 31; 479-83
6.. Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T, Plasmatherapy in atypical hemolytic uremic syndrome: Semin Thromb Hemost, 2010; 36; 673-81
7.. Zuber J, Fakhouri F, Roumenina LT, Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies: Nat Rev Nephrol, 2012; 8; 643-57
8.. Arnold DM, Patriquin CJ, Nazy I, Thrombotic microangiopathies: A general approach to diagnosis and management: CMAJ, 2017; 189(4); E153
9.. Laurence J, Haller H, Mannucci PM, Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis: Clin Adv Hematol Oncol, 2016; 14(Suppl. 11); 2-15
10.. Asif A, Nayer A, Haas CS, Atypical hemolytic uremic syndrome in the setting of complement-amplifying conditions: Case reports and a review of the evidence for treatment with eculizumab: J Nephrol, 2017; 30; 347-62
11.. Shibagaki Y, Fujita T, Thrombotic microangiopathy in malignant hypertension and hemolytic uremic syndrome (HUS)/thrombotic thrombocytopenic purpura (TTP): Can we differentiate one from the other?: Hypertens Res, 2005; 28(1); 89-95
12.. Totina A, Iorember F, El-Dahr SS, Yosypiv I, Atypical hemolytic-uremic syndrome in a child presenting with malignant hypertension: Clin Pediatr (Phila), 2013; 52; 183-86
13.. Timmermans SAMEG, Abdul-Hamid MA, Vanderlocht J, Patients with hypertension-associated thrombotic microangiopathy may present with complement abnormalities: Kidney Int, 2017; 91; 1420-25
14.. Larsen CP, Wilson JD, Best-Rocha A, Genetic testing of complement and coagulation pathways in patients with severe hypertension and renal microangiopathy: Modern Pathology, 2018; 31; 488-94
15.. Maga TK, Nishimura CJ, Weaver AE, Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome: Hum Mutat, 2010; 31; E1445-60
16.. Noris M, Caprioli J, Bresin E, Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype: Clin J Am Soc Nephrol, 2010; 5; 1844-59
17.. Michon B, Moghrabi A, Winikoff R, Complications of apheresis in children: Transfusion, 2007; 47(10); 1837-42
18.. Legendre CM, Licht C, Muus P, Terminal complement inhibitor Eculizumab in atypical hemolytic-uremic syndrome: N Engl J Med, 2013; 368; 2169-81
Tables
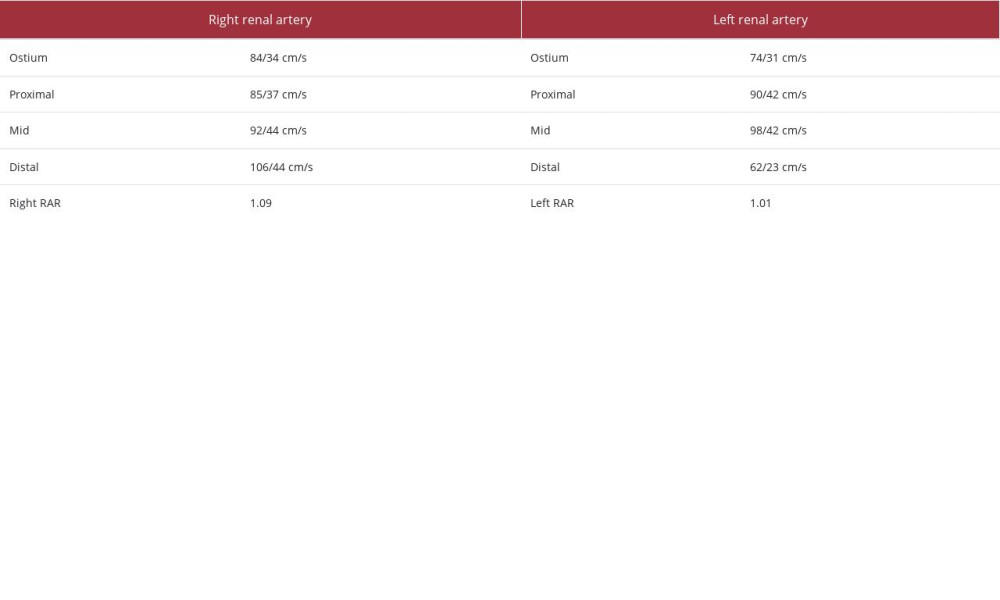 Table 1.. Renal artery Doppler ultrasound showing normal flow through the renal arteries.
Table 1.. Renal artery Doppler ultrasound showing normal flow through the renal arteries.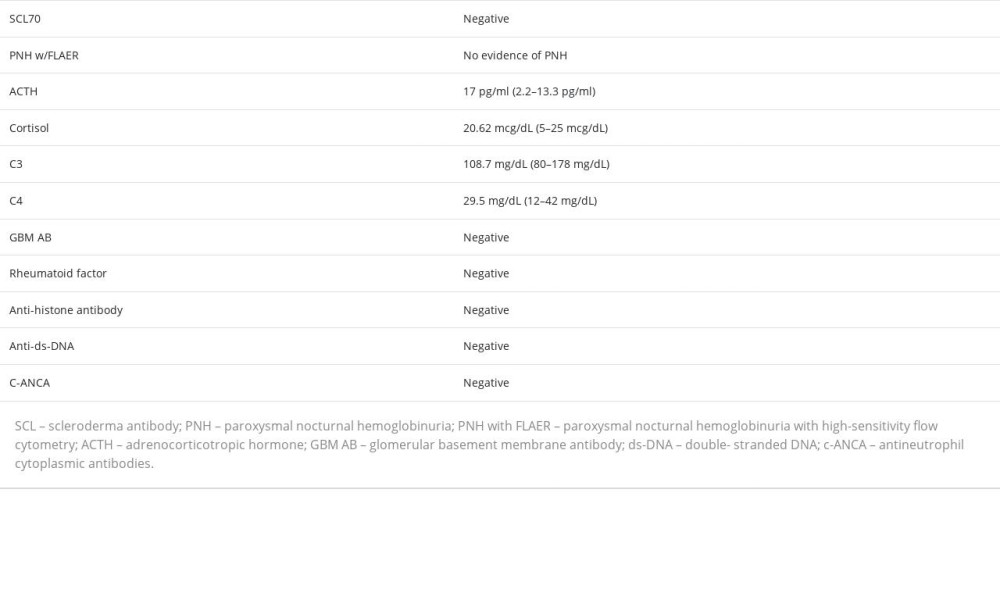 Table 2.. Secondary work-up for hypertension and TMA.
Table 2.. Secondary work-up for hypertension and TMA.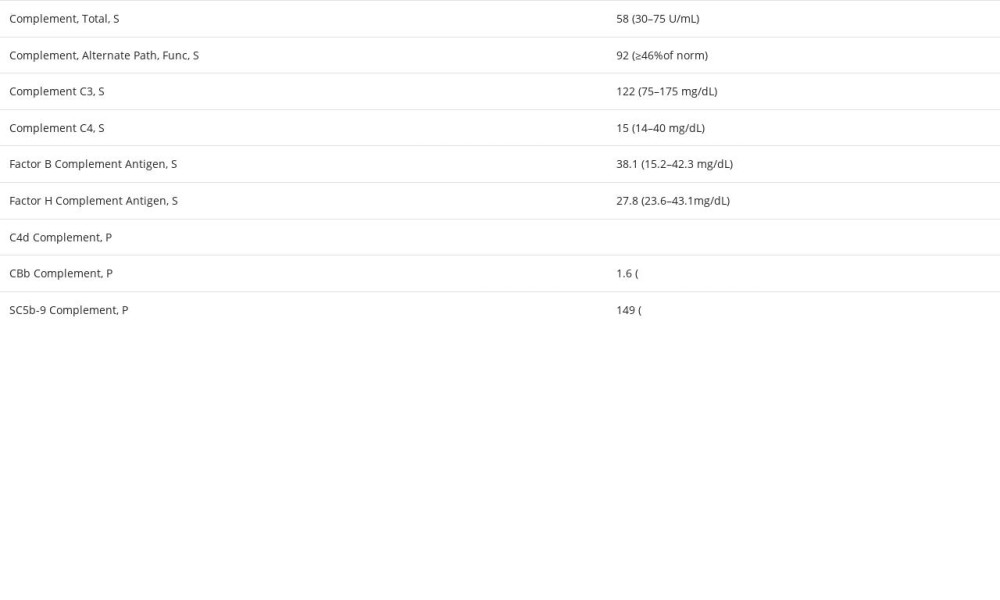 Table 3.. Complement-mediated TMA panel.Table 1.. Renal artery Doppler ultrasound showing normal flow through the renal arteries.Table 2.. Secondary work-up for hypertension and TMA.Table 3.. Complement-mediated TMA panel.
Table 3.. Complement-mediated TMA panel.Table 1.. Renal artery Doppler ultrasound showing normal flow through the renal arteries.Table 2.. Secondary work-up for hypertension and TMA.Table 3.. Complement-mediated TMA panel. In Press
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953173
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.953192
Case report 
Am J Case Rep In Press; DOI: 10.12659/AJCR.952818
Case report
Am J Case Rep In Press; DOI: 10.12659/AJCR.953608
Most Viewed Current Articles
07 Dec 2021 : Case report
22,364,578
DOI :10.12659/AJCR.934347
Am J Case Rep 2021; 22:e934347
06 Dec 2021 : Case report  174,245
174,245
DOI :10.12659/AJCR.934406
Am J Case Rep 2021; 22:e934406
21 Jun 2024 : Case report
119,744
DOI :10.12659/AJCR.944371
Am J Case Rep 2024; 25:e944371
07 Mar 2024 : Case report
64,648
DOI :10.12659/AJCR.943133
Am J Case Rep 2024; 25:e943133